Synthesis of the Telomeric C-Strand. A Review
C. M. Price1
1Department of Chemistry and Department of Biochemistry, University of Nebraska, Lincoln, NE 68588, USA; fax: (402) 472-3518; E-mail: cprice@unlinfo.unl.edu
Submitted August 1, 1997.
Fill-in synthesis of the C-strand DNA plays a critical role in determining telomere length.
KEY WORDS: telomere, DNA replication.
The telomeric DNA of many organisms consists of a simple tandemly repeated sequence which is G-rich on the strand that extends towards the 3´ terminus of the chromosome (the G-strand) and C-rich on the complementary 5´ strand (the C-strand) [1]. It is now well established that the telomeric G-strand is synthesized by the enzyme telomerase and the mechanism of telomerase action has been studied extensively [2]. However, relatively little effort has gone into examining how the complementary C-strand is synthesized. This article explores the extent to which C-strand synthesis is required both during regular DNA replication and during more specialized events such as the new telomere synthesis that can take place following chromosome fragmentation. The evidence for the involvement of DNA polymerase alpha (polalpha)/primase in C-strand synthesis is then discussed.
Telomerase and the End-Replication Problem
One of the main reasons why linear chromosomes have telomeres is to circumvent the loss of DNA that takes place when the 5´ terminus of a chromosome is replicated [3, 4]. This loss of 5´ terminal sequence occurs because all known DNA polymerases require a pre-existing primer molecule in order to initiate DNA synthesis. The primer is usually a short RNA molecule that is generated by DNA primase, extended by DNA polymerase, and later removed by nucleases [5]. Removal of the primer results in the daughter strand being 8-16 nucleotides shorter than the parental strand.
Telomerase solves this "end-replication" problem by adding DNA to the end of a chromosome by a process that is independent of the regular replication machinery. As described elsewhere in this series of reviews, telomerase is an unusual reverse transcriptase that has an RNA molecule as an integral component of the enzyme [2]. The RNA molecule has a short template region that is complementary to ~1.5 telomeric repeats. During addition of new telomeric DNA, the partial repeat aligns with the end of the chromosome and the full repeat is copied onto the end of the pre-existing DNA. To generate multiple repeats, the enzyme then translocates to the new chromosome terminus, realigns with the template region and the process is repeated.
The Need for C-Strand Synthesis
Synthesis of new telomeric C-strands can be separated into two mechanistically different reactions: 1) the replication of preexisting telomeric DNA (C-strand replication), and 2) the generation of double-stranded telomeric DNA from stretches of single-stranded G-repeats such as those laid down by telomerase (C-strand fill-in). Since telomerase is only needed to maintain the DNA at the extreme terminus of the chromosome, most preexisting telomeric DNA is replicated by normal semiconservative replication using the standard replication machinery. Except in rare cases where the telomere may act as an origin [6, 7], this means that the bulk of the telomeric DNA will be generated by leading and lagging strand synthesis from a bidirectional replication fork [8, 9]. Thus, in organisms where the telomeric DNA is longer than ~1 kb, much of the C-strand will be generated as a series of Okazaki fragments by the normal lagging strand replication enzymes [10]. In contrast, a standard replication fork can not be used to generate new C-strand opposite regions of telomerase generated G-strand. However, as discussed below, it is likely that this C-strand "fill-in" is still achieved by polalpha/primase and possibly PCNA and pol. or polepsilon.
The extent to which C-strand fill-in is required probably varies from species to species. Extensive new C-strand synthesis is clearly needed in those organisms which undergo developmentally regulated new telomere addition. A lesser amount will also be required in cells where telomere length regulation has been perturbed and the telomeres are being elongated. However, it is still unclear how much C-strand fill-in is required during DNA replication. Since telomere length does not normally change substantially as cells proliferate, it has been suggested that telomerase may add only a small number of repeats at each round of replication. Thus, only a very limited amount of C-strand fill-in might be required. However, the recent discovery that long stretches of single-stranded G-repeats can be generated by a nuclease rather than telomerase [11] provides evidence that extensive C-strand fill-in may be necessary in at least some organisms.
C-Strand Synthesis during New Telomere Addition
New telomere synthesis occurs as a developmentally regulated process in the life cycle of a number of different organisms such as ciliates, nematodes, certain insects, and crustaceans [12-14]. It also occurs as a rare event in many different organisms (including humans and yeast) when a chromosome is broken and then healed [15, 16]. During new telomere synthesis an entire telomere is added onto the end of a broken chromosome which lacks any pre-existing telomeric sequence. Thus, not only long stretches of G-strand, but also C-strand must be generated.
Developmentally Programmed Telomere Addition in Ciliates. New telomere synthesis takes place in ciliated protozoa during the sexual phase of the life cycle when the new macronucleus is being generated [12]. Ciliates have two types of nuclei: the germline micronucleus which is transcriptionally inert, and the vegetative macronucleus which is transcriptionally active. The macronucleus is generated from a copy of the micronucleus after conjugation and fertilization, during a process that involves fragmentation of the micronuclear chromosomes and addition of telomeres to the newly generated ends [12, 17]. The extent of this fragmentation and new telomere synthesis varies between different ciliate species. In Tetrahymena the five pairs of micronuclear chromosomes are fragmented into ~200 macronuclear chromosomes with an average size of 600 kb. In the hypotrichs Euplotes and Oxytricha the micronuclear chromosomes become so heavily fragmented that each individual gene is released as a separate DNA molecule. These gene-sized molecules have telomeres added to either end so each macronucleus ends up with ~2·107 separate DNA molecules and ~4·107 telomeres.
The first step in new telomere synthesis is the generation of the G-strand by telomerase [18]. However, new telomeres that have long stretches of single-stranded telomeric DNA cannot normally be detected in developing cells (J. Vermeesch and C. Price, unpublished observations, and [19]), so the C-strand must be synthesized soon after G-strand addition. In Tetrahymena, mature telomeres of 0.3-0.4 kb can be detected on the newly formed macronuclear DNA molecules within 24 h after mating [20]. This means that long stretches of both G- and C-strand telomeric DNA must be generated quite rapidly. In Euplotes, full length telomeres are generated over a period of a few hours [21]. The newly synthesized G-strands are rather heterogeneous in length but the majority are 95-100 nucleotides long [22]. In contrast, most of the C-strands are exactly 84 nucleotides long. Thus, new telomere addition involves not only synthesis of the telomeric G- and C-strands but also tight regulation of C-strand length.
Developmentally Programmed Telomere Addition in Nematodes. In nematodes chromosome fragmentation and new telomere addition take place as part of a developmentally regulated process called chromatin diminution [13]. Chromatin diminution occurs during the first 2-8 cleavage divisions in a wide variety of nematodes and is a process that allows the presomatic cells to eliminate large amounts of satellite DNA as well as germline specific sequences. The events leading to chromatin diminution have been most closely studied in the nematodes Parascaris univalens and Ascaris lumbricoides. In P. univalens, the central portion of the germline chromosomes break up into many smaller chromosomes which are retained during the next mitotic division. The terminal regions of germline chromosomes, which contain the satellite DNA, do not attach to the mitotic spindle and are lost from the nucleus. In A. lumbricoides it is not know whether the central region of the germline chromosomes become fragmented; however, ~25% of the germline DNA is discarded when the heterochromatic termini of the chromosomes break off and fail to attach to the mitotic spindle. In both species, new telomeres are added to the ends of the fragmented chromosomes [23, 24]. In A. lumbricoides fragmentation takes place within specific regions of the chromosome termed CBR (Chromosome Breakage Regions) [23]. Telomere addition can then occur at multiple sites along the CBR. During telomere addition 2-4 kb of the sequence TAGGC·ATCCG is added to the end of the broken chromosome. Thus, long stretches of both telomeric G- and C-strand must be synthesized.
Spontaneous Telomere Addition Following Chromosome Breakage. Unscheduled chromosome breakage is not necessarily a lethal event because a broken chromosome may be stabilized by the addition of telomeric DNA to the break point. The healing of broken chromosomes by de novo telomere addition has been observed in many different organisms including plants, humans, yeast, and the malaria parasite Plasmodium falciparum [15, 16, 25-27]. Spontaneous chromosome breakage and healing seems to be particularly common in certain plants and in Plasmodium. Many wheat and barley stocks have been isolated which exhibit terminal deletions that have been healed by the addition of the plant telomere sequence T3AG3·A3TC3 [26, 28]. Likewise, field isolates of Plasmodium exhibit considerable polymorphism in chromosome size as a result of terminal deletions and rearrangements [27]. When maintained in culture, clinical isolates undergo frequent chromosome breakage and loss of the antigen genes which are located in the subtelomeric region [29]. In P. falciparum the breakpoints are healed by the addition of 1.5-2.5 kb of telomeric DNA [30].
Healing of broken chromosomes appears to be less common in mammals, but has been clearly documented in patients with alpha-thalassemia [15, 31]. In these patients, chromosome breakage occurred within the alpha-globin gene cluster on the distal region of chromosome 16q. Sequencing of the terminal restriction fragment revealed that telomeric sequence had been added directly to the broken end.
Efficient chromosome healing only seems to occur in cells that express telomerase. For example, in plants healing only takes place in the embryo, and not the telomerase deficient endosperm ([25] and D. Shippen, personal communication). Likewise, in mammalian cells healing of artificially induced chromosome breaks was only observed in transformed cells and stem cells but not in telomerase negative primary human fibroblasts [32]. Sequence analysis of the site of telomere addition has revealed that in many cases the break site contains a few nucleotides that are complementary to the template region of telomerase RNA [16, 31, 33]. These observations suggest that healing is initiated by telomerase adding G-strand repeats directly onto the end of the broken chromosome. Presumably C-strand synthesis follows soon afterwards.
C-Strand Synthesis during Telomere Elongation
Synthesis of additional double-stranded telomeric DNA and, hence, the telomeric C-strand occurs when telomeres become longer. Elongation of telomeres has been observed in Tetrahymena and trypanosomes under certain growth conditions [20, 34], as well as in various mutant yeast strains that have defects in factors needed for telomere maintenance. The elongation of trypanosome telomeres occurs when the cells are grown continuously in host organisms such as mice or rats [34]. Under these conditions the telomeres grow at a rate of 7-10 bp/generation for hundreds of generations. However, the telomeres do not grow to an infinite length because sudden large deletions eventually cause telomere shortening. Growth of Tetrahymena telomeres is also observed when the cells are kept in continuous culture [20]. The growth rate is 3-10 bp/generation. Again growth does not continue indefinitely because the cultures are eventually taken over by mutant cells with short telomeres ([20] and E. Henderson, personal communication). While additional C-strand synthesis is clearly required as trypanosome and Tetrahymena telomeres grow, the amount of new C-strand that is generated per generation is much smaller than the large scale synthesis that occurs during new telomere formation.
Elongation of yeast telomeres has been observed in quite a wide variety of mutant strains. For example, mutations in the telomere-binding proteins RAP1, RIF1, and Taz1 cause elongation, as do mutations in polymerase alpha, replication factor C, and the helicase PIF1 [35-38]. Certain mutations in the template region of yeast telomerase also cause telomere elongation [39]. The growth of yeast telomeres in the telomerase and telomere-binding protein mutants can probably be explained in terms of altered chromatin structure [36, 40]. It is thought that the mutant telomere binding protein or the mutant telomeric DNA sequence (which results from the telomerase template mutation) prevents the telomeric DNA from being packaged into the normal compact telomeric chromatin structure. This would then allow telomerase greater access to the DNA terminus, and hence the opportunity to add more G-strand repeats. Subsequent synthesis of the complementary C-strand repeats would result in a net increase in telomere length. The amount of new telomeric DNA that is synthesized per generation varies for each mutation; however, in some cases (e.g., in the telomerase template mutants) it can be on the order of hundreds of base pairs.
C-Strand Fill-in During DNA Replication in Yeast
The terminal regions of yeast chromosomes are replicated late in S-phase by conventional bidirectional replication [8, 35, 41]. Replication origins are located internally to the subtelomeric DNA and the resulting replication forks duplicate the subtelomeric sequences and probably also part of the telomeric DNA [9, 41]. However, the most terminal portion of the telomeric DNA is maintained by a separate mechanism that seems to involve the action of not only telomerase but also a 5´-exonuclease [11]. Information about the mechanism of yeast telomere replication has been obtained by analyzing the structure of the telomeric G-strand during different stages of the cell cycle [9, 42]. This analysis revealed that long single-stranded tails appear on the telomeric G-strand immediately following passage of the replication fork to the end of the chromosome. The long G-tails are at least 30 nucleotides in length and are probably in the 30-150 nucleotide range. They exist only transiently during the cell cycle and are lost as the cells enter G2/M.
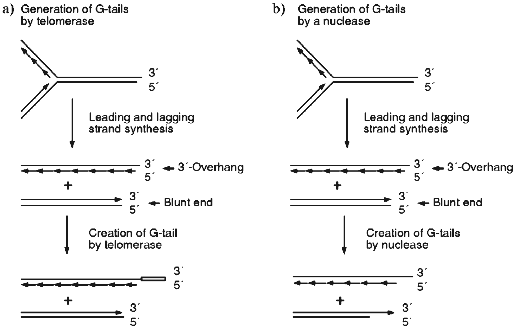
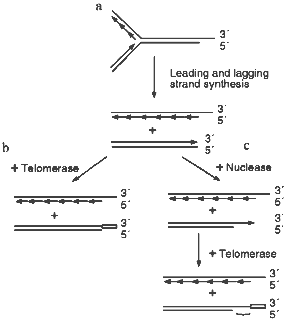
The most obvious way to generate the G-strand tails is via that action of telomerase. However, careful analysis of the G-strand structure of a linear YAC revealed that the G-tails are present at both ends of the chromosome [43]. This finding indicated that telomerase is unlikely to be the sole mechanism for generating the G-tails because bidirectional replication of a yeast chromosome should result in a DNA molecule that has one blunt end and one end with a short 3´-overhang (see Fig. 1a). Only the end with the 3´-overhang should be extended by telomerase because the enzyme does not use double-stranded DNA as a substrate [44, 45]. Subsequent experiments demonstrated that the G-tails are generated even in cells that lacked telomerase [11, 43]. Thus, the tails must be generated by a telomerase independent mechanism; this probably involves an exonuclease which removes the 5´ terminal portion of the C-strand (Fig. 1b). Removal of a region of the C-strand could serve both to make the newly generated leading strand a substrate for telomerase (Fig. 1, c and d) and to generate short (<30 nucleotides) G-strand overhangs that may be necessary for the binding of telomere end-binding proteins.
Since the long G-tails always disappear at the end of S-phase, they must either be removed by a nuclease (Fig. 1c) or converted to double-stranded DNA by filling-in the C-strand (Fig. 1d). It is unlikely that the tails are simply removed by a nuclease since this would lead to a net decrease in telomere length. Half of the telomeres would decrease in length by 30-150 nucleotides/generation if one G-tail is generated by C-strand degradation and the other is generated by telomerase, while all the telomeres would decrease by this amount if both G-tails are generated by the endonuclease. Wild type yeast do not show a net decrease in telomere length following log phase growth [9, 42, 46, 47] and even telomerase-deficient mutants exhibit a much more gradual telomere shortening (~3 bp/generation [48]). Thus, the disappearance of the G-tails at the end of S-phase is most likely to be due to fill-in of the telomeric C-strand. As discussed below, this is probably achieved by the regular DNA replication machinery.Fig. 1. Mechanisms for generating and removing G-strand tails from yeast telomeres. a, b) The parental molecule is replicated by leading and lagging strand synthesis from a bidirectional replication fork. This generates one telomere with a 3´-overhang and one with a blunt end. Leading strand synthesis is shown as one long arrow, lagging strand synthesis is represented by a series of short arrows. a) Telomerase can only add extra repeats onto the molecule with the 3´-overhang. The open box represents the new G-strand DNA made by telomerase. b) An exonuclease removes part of the C-strand from the two replication intermediates. Thus, both telomeres are now substrates for telomerase. c, d) Telomerase adds repeats onto the bottom molecule created as shown in (b), therefore the final G-strand length is the same as that of the parental molecule. c) The whole 3´-overhang is removed by a nuclease. This leads to net telomere shortening. d) Most of the G-strand overhang is converted to double-stranded DNA by C-strand fill-in. The resulting telomere is the same length as the parental molecule.
Is C-Strand Fill-in Required during DNA Replication in Higher Eukaryotes?
Cells from higher eukaryotes can be divided into two classes; those that have a mechanism for solving the end replication problem and those that do not [49, 50]. Cells that can solve the end replication problem include germline cells, tumor cells, and some somatic cells, e.g., from some mouse tissue. These cells either contain active telomerase or have developed another mechanism, termed ALT, for telomere maintenance [51, 52]. ALT stands for alternate telomere maintenance and is probably recombination based. Cells that lack a mechanism for solving the end replication problem include normal human somatic cells and some mouse tissue, e.g., brain [50, 51]. These cells lack significant levels of telomerase and display progressive telomere shortening following DNA replication. As previously discussed, the bulk of the telomeric DNA in both classes of cells is probably replicated by regular leading and lagging strand synthesis. In cells that lack telomerase or ALT, this is probably the only mechanism for generating new C-strand DNA. Cells that have telomerase may also generate new C-strand by filling-in opposite regions of single-stranded G-repeats. However, the extent to which this takes place is currently unclear.
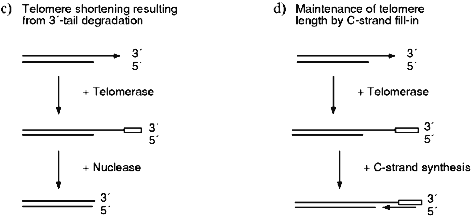
Several recent studies have demonstrated that the telomeres from human cells have long G-strand overhangs ([53, 54] and W. Wright, personal communication). These overhangs have a median length of 150-200 nucleotides and they are present in both normal somatic cells as well as transformed cell lines. Unlike the G-strand overhangs observed in yeast, the human overhangs seem be present throughout the cell cycle as they can be detected in both quiescent and actively dividing cells. However, it is still unclear whether they are present on one or both ends of the chromosome. If the overhangs are present on only one end, they probably result from failure of the replication machinery to lay down the last Okazaki fragment during lagging strand (C-strand) synthesis (Fig. 2a). However, if they are present on both ends of the chromosome, the second overhang is probably generated by a 5´-endonuclease as occurs in yeast (Fig. 2c).
Since cells that lack telomerase and ALT have no way to compensate for the loss of C-stand DNA, telomere shortening is a predicted outcome of DNA replication (Fig. 2a). However, no such decrease in telomere length is observed in cells that have telomerase, so the loss of C-strand from one or both ends of the chromosome must be compensated for by the addition of new G-strand by telomerase. If the G-strand overhangs are only generated by incomplete DNA replication (Fig. 2a), the simple addition of 150-200 nucleotides of G-strand to the blunt-ended telomere might be sufficient to complete telomere replication (Fig. 2b). Since the overhangs appear to remain throughout the cell cycle, fill-in of the C-strand may not be required. Obviously, for this mechanism to be feasible the two strands of the blunt-ended molecule would have to be somehow separated so that the G-strand can become a substrate for telomerase. In contrast, if the G-strand overhangs are generated by both incomplete DNA replication and via nuclease action (Fig. 2c), C-strand synthesis will be required to fill-in the region of single-strand generated by the nuclease. If no C-strand fill-in were to occur, the G-strand overhangs would be much longer than those that have been detected experimentally.Fig. 2. Replication of human telomeres. a) The parental molecule is replicated by leading and lagging strand synthesis. Failure to lay down the final full length Okazaki fragment during lagging strand synthesis results in 50% of the telomeres having G-strand overhangs that are on average half the length of an Okazaki fragment (i.e., 100-200 nucleotides). The remaining telomeres are blunt-ended. b) The G-strand of the blunt-ended molecule is extended to the length of the parental G-strand by telomerase. This results in identical daughter and parental molecules. c) A G-strand overhang is generated on the blunt-ended telomere by a nuclease, the newly synthesized G-strand is then extended to the length of the parental G-strand by telomerase. This results in a G-strand overhang that is much longer than those observed experimentally. Thus, the region marked with a bracket must be converted to duplex DNA by C-strand fill-in.
The Mechanism of C-Strand Fill-in
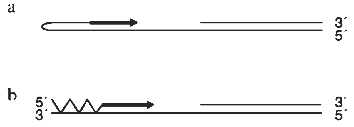
There are two obvious mechanisms for generating C-strand DNA opposite a single-stranded region of G-repeats. These are by priming from a G-strand fold-back structure and by using polalpha/primase to initiate synthesis (Fig. 3). It is well established that telomeric G-strand DNA has the capacity to form a variety of unusual structures including intramolecular quadruplexes and duplexes that involve non-Watson--Crick base pairing [55]. Consequently, it had been suggested that C-strand synthesis could be primed by the G-strand folding back to form a hairpin with a free 3´-OH [14, 56]. However, it is not apparent that there are several problems with this suggestion. The first problem is that a foldback structure is likely to be a poor substrate for DNA polymerase because it cannot form Watson--Crick base pairs. Indeed, the non-canonical base pairs will probably be recognized as mismatches and hence be removed by the proofreading endonuclease activity. The second problem is that the proteins from ciliated protozoa which are known to bind to the G-strand overhang recognize single-stranded DNA and can not bind any form of a duplex or quadruplex structure ([57] and Steven and Schultz, personal communication). Thus, the G-strand DNA is not likely to exist as a fold back structure unless the telomere-binding protein is dissociated.
The most likely mechanism for generating new C-strand DNA is through the activity of the enzymes that are normally used to generate lagging strand DNA, i.e., polalpha/primase in conjunction with PCNA and pol. and/or polepsilon. While primer synthesis and extension would not be carried out as part of coupled leading and lagging strand synthesis at a replication fork, this is unlikely to be a problem as uncoupling of bidirectional replication is observed during DNA repair when pol. or polepsilon synthesize a repair patch [58, 59].Fig. 3. Methods for priming synthesis of the telomeric C-strand. a) The G-strand forms a fold-back structure that can be extended by DNA polymerase. b) Polalpha/primase generates an RNA primer that is then extended by polalpha.
Some evidence that the lagging strand replication machinery is responsible for C-strand fill-in has been obtained from the analysis of telomere length in yeast mutants. Carson and Hartwell were the first to demonstrate that a mutation in polalpha resulted in an increase in telomere length when S. cerevisiae were grown at the semi-permissive temperature (Carson and Hartwell, 1985). It has since been demonstrated that a variety of polalpha alleles as well as some pol. and replication factor C alleles cause telomere elongation ([38] and T. Petes, personal communication). A similar effect was observed in the fission yeast S. pombe with polalpha mutations (T. Cech, personal communication). While the effect of the polymerase and replication factor C mutations suggest that these proteins are involved in telomere metabolism, it is also possible that the mutations merely cause a secondary effect on telomere length as a result of a general replication defect. However, evidence that an alteration in polalpha activity causes a direct effect on telomere metabolism has recently been obtained from experiments with the ciliate Euplotes crassus [19].
As discussed earlier, Euplotes cells contain literally millions of telomeres that are generated by a multi-step process during the sexual stage of the life cycle [12]. Since no general DNA replication occurs during the new telomere synthesis, Euplotes provides a unique opportunity to study the role of DNA polymerase in telomeric C-strand synthesis without the complication of simultaneous DNA replication. When Euplotes cells are allowed to undergo new telomere synthesis in the presence of the DNA polymerase inhibitor aphidicolin, a dramatic effect is observed on the length of both the telomeric G- and C-strands [19]. If sufficient aphidicolin is used to only partially inhibit polalpha, the median length of the C-strands remains unchanged. However, the overall length distribution becomes much more heterogeneous so the majority of the C-strands are no longer precisely 84 nucleotides long. Since the aphidicolin causes this change in C-strand length at a time when no general DNA replication is taking place, the above observation provides good evidence that DNA polalpha is involved in C-strand synthesis.
Interestingly, the effect of aphidicolin on G-length is quite different from the effect on the C-strands. Following aphidicolin treatment the G-strands generally become longer, with some reaching up to four times the normal length. This increase in length is not caused by the aphidicolin stimulating telomerase activity, as the drug has no measurable affect on the enzyme. The unexpected finding that aphidicolin causes a change in not only C- but also G-strand length indicates that synthesis of the telomeric G- and C-strands are coordinately regulated. Since aphidicolin is such a specific inhibitor of DNA polalpha and pol., this coordinate regulation is most likely mediated by DNA polymerase. One way to achieve the coordinate regulation of G- and C-strand length would be by having the initiation of C-strand synthesis (by polalpha/primase) prevent telomerase from adding more T4G4 repeats to the G-strand. When polalpha is partially inhibited by aphidicolin, telomerase would have more time to elongate the G-strand before being inhibited by the initiation of C-strand synthesis.
Analysis of the telomeres from a number of different organisms has revealed that synthesis of telomeric C-strand DNA occurs both during regular bidirectional DNA replication and as a result of a separate "fill-in" reaction that converts regions of single-stranded G-repeats to double-stranded telomeric DNA. The latter fill-in reaction appears to be absolutely required in a number of places in nature. For example, new telomere addition requires that the complementary C-strand be synthesized once telomerase has laid down the G-strand. Likewise, synthesis of the complementary C-strand is required to achieve net telomere elongation. The requirement for C-strand fill-in during DNA replication is intriguing and somewhat unexpected. While one might have predicted that little C-strand fill-in would be required because relatively little change in telomere length occurs, it appears that in at least some organisms telomere replication is more complex than was first thought. In these organisms both telomerase and a nuclease(s) act on the telomere. This results in the generation of long stretches of single-strand G-repeats which must later be converted to double-strand DNA by the C-strand fill-in reaction.
Fill-in synthesis of C-strand DNA clearly plays a critical role in determining telomere length because it complements the activity of other enzymes (telomerase or nuclease) which directly alter the length of the telomeric G- or C-strand. However, in addition to this passive role in telomere length regulation, C-strand synthesis may be actively involved in determining how many repeats telomerase can add to a DNA terminus. The most clear-cut example of this active role in length regulation is seen during new telomere addition in Euplotes, where alteration of C-strand synthesis causes a change in G-strand length. While the mechanism by which the enzymes responsible for C-strand synthesis exert their effect on G-strand length is unknown, it could be that polalpha/primase actually forms a complex with telomerase and this complex regulates telomerase activity.
The other case where there is evidence that C-strand synthesis may directly regulate telomere length is in S. cerevisiae, where mutations in the lagging strand replication machinery have been shown to cause an increase in the length of the telomeric DNA. This effect could be the result of the lagging strand replication machinery forming a complex with telomerase and thus directly regulating telomerase activity. Alternatively, the mutations in the lagging strand enzymes might affect telomerase activity less directly. For example, the rate at which the G-strand overhangs are converted to duplex DNA could regulate the length of time that telomerase has access to the DNA terminus. Once the G-strand is converted to duplex DNA, binding of the telomere protein RAP1 may render the DNA terminus inaccessible to telomerase. Thus, if synthesis of the C-strand were to occur less efficiently, telomerase would have more time to added repeats to the G-strand. This would lead to an increase in telomere length.
LITERATURE CITED
1.Henderson, E. (1995) in Telomeres
(Blackburn, E., and Greider, C., eds.) Cold Spring Harbor Laboratory
Press, New York, pp. 11-34.
2.Greider, C. W. (1995) in Telomeres
(Blackburn, E., and Greider, C., eds.) Cold Spring Harbor Laboratory
Press, New York, pp. 35-68.
3.Olovnikov, A. M. (1971) Dokl. Biochem.,
201, 685-691.
4.Watson, J. D. (1972) Nature New Biol.,
239, 197-201.
5.Kornberg, A. (1992) DNA Replication, W. H.
Freeman Press.
6.Zahler, A. M., and Prescott, D. M. (1989)
Nucleic Acids Res., 17, 6299-6317.
7.Skopp, R., Wang, W., and Price, C. (1996)
Chromosoma, 105, 82-91.
8.Ferguson, B., and Fangman, W. L. (1992)
Cell, 68, 333-339.
9.Wellinger, R. J., Wolf, A. J., and Zakian, V. A.
(1993) Mol. Cell Biol., 13, 4057-4065.
10.Reveal, P. M., Henkels, K. M., and Turchi, J. J.
(1997) J. Biol. Chem., 272, 11678-11681.
11.Dionne, I., and Wellinger, R. J. (1996) Proc.
Natl. Acad. Sci. USA, 93, 13902-13907.
12.Prescott, D. M. (1994) Microbial Rev.,
58, 233-267.
13.Tobler, H., Etter, A., and Muller, F. (1992)
Trends Genet., 8, 427-432.
14.Blackburn, E. H., and Szostak, J. W. (1984)
Ann. Rev. Biochem., 53, 163-194.
15.Wilkie, A. O. M., Lamb, J., Harris, P. C.,
Finney, R. D., and Higgs, D. R. (1990) Nature, 346,
868-871.
16.Kramer, K. M., and Haber, J. E. (1993) Genes
Dev., 7, 2345-2356.
17.Blackburn, E. H. (1995) in Telomeres
(Blackburn, E., and Greider, C., eds.) Cold Spring Harbor Laboratory
Press, New York, pp. 193-218.
18.Yu, G. L., and Blackburn, E. (1991) Cell,
67, 823-832.
19.Fan, X., and Price, C. M. (1997), submitted.
20.Larson, D. D., Spangler, E. A., and Blackburn, E.
H. (1987) Cell, 50, 477-483.
21.Roth, M., and Prescott, D. M. (1985) Cell,
41, 411-417.
22.Vermeesch, J. R., and Price, C. M. (1994) Mol.
Cell Biol., 14, 554-566.
23.Muller, F., Wichy, C., Spicher, A., and Tobler,
H. (1991) Cell, 67, 815-822.
24.Zetka, M. C., and Muller, F. (1996) Sem. Cell
Dev. Biol., 7, 59-64.
25.McClintock, B. (1941) Genetics,
26, 234-281.
26.Werner, J. E., Kota, R. S., and Gill, B. S.
(1992) Genome, 35, 844-848.
27.Scherf, A. (1996) Sem. Cell Dev. Biol.,
7, 49-57.
28.Wang, S., Lapitan, N. L. V., Roder, M., and
Tsuchiya, T. (1992) Genome, 35, 975-980.
29.Scherf, A., and Mattei, D. (1992) Nucleic
Acids Res., 7, 1491-1496.
30.Scherf, A., Carter, R., Petersen, C., Alano, P.,
Nelson, R., Aikawa, M., Mattei, D., Pereira Da Silva, L., and Leech, J.
(1992) EMBO J., 11, 2293-2301.
31.Flint, J., Craddock, C. F., Villegas, A.,
Bentley, D. P., Williams, H. J., Galanello, R., Cao, A., Wood, W. G.,
Ayyub, H., and Higgs, D. R. (1994) Am. J. Hum. Genet.,
55, 505-512.
32.Barnett, M. A., Buckle, V. J., Evans, E. P.,
Porter, A. C. G., Rout, D., Smith, A. G., and Brown, W. R. A. (1993)
Nucleic Acids Res., 21, 27-36.
33.Melek, M., and Shippen, D. E. (1996)
Bioessays, 18, 301-308.
34.Bernards, A., Michels, P. A. M., Lincke, C. R.,
and Borst, P. (1983) Nature, 303, 592-597.
35.Zakian, V. A. (1995) in Telomeres
(Blackburn, E., and Greider, C., eds.) Cold Spring Harbor Laboratory
Press, New York, pp. 107-138.
36.Krauskopf, A., and Blackburn, E. (1996)
Nature, 383, 354-357.
37.Cooper, J. P., Nimmo, E. R., Allshire, R. C., and
Cech, T. R. (1997) Nature, 385, 744-747.
38.Adams, A. K., and Holm, C. (1996) Mol. Cell
Biol., 16, 4614-4620.
39.McEachern, M. J., and Blackburn, E. H. (1995)
Nature, 376, 403-409.
40.Marcand, S., Gilson, E., and Shore, D. (1997)
Science, 275, 986-990.
41.Ferguson, B. M., Brewer, B. J., Reynolds, A. E.,
and Fangman, W. L. (1991) Cell, 65, 507-515.
42.Wellinger, R. J., Wolf, A. J., and Zakian, V. A.
(1993) Cell, 72, 51-60.
43.Wellinger, R. J., Ethier, K., Labrecque, P., and
Zakian, V. A. (1996) Cell, 85, 423-433.
44.Henderson, E., and Blackburn, E. (1989) Mol.
Cell Biol., 9, 345-348.
45.Lingner, J., and Cech, T. R. (1996) Proc.
Natl. Acad. Sci. USA, 93, 10712-10717.
46.Shampay, J., and Blackburn, E. H. (1988) Proc.
Natl. Acad. Sci. USA, 85, 534-538.
47.D'Mello, N. P., and Jazwinski, S. M. (1991) J.
Bacteriol., 173, 6709-6713.
48.Singer, M. S., and Gottschling, D. E. (1994)
Science, 266, 404-409.
49.De Lange, T. (1995) in Telomeres
(Blackburn, E., and Greider, C., eds.) Cold Spring Harbor Laboratory
Press, New York, pp. 295-338.
50.Harley, C. B. (1995) in Telomeres
(Blackburn, E., and Greider, C., eds.) Cold Spring Harbor Laboratory
Press, New York, pp. 247-264.
51.Bacchetti, S. (1996) Sem. Cell Dev. Biol.,
7, 31-39.
52.Bryan, T. M., and Reddel, R. R. (1997) Eur. J.
Cancer, 35, 767-773.
53.Makarov, V. L., Hirose, Y., and Langmore, J. P.
(1997) Cell, 88, 657-666.
54.McElligott, R., and Wenninger, R. J. (1997)
EMBO J., in press.
55.Williamson, J. R. (1994) Ann. Rev. Biophys.
Biomol. Struct., 23, 703-730.
56.Zakian, V. A. (1989) Ann. Rev. Genet.,
23, 579-604.
57.Zahler, A. M., Wiliamson, J. R., Cech, T. R., and
Prescott, D. M. (1991) Nature, 350, 718-720.
58.Sancar, A. (1996) Ann. Rev. Biochem.,
65, 43-82.