Saccharomyces cerevisiae Telomeres. A Review
F. E. Pryde1 and E. J. Louis1,2
1Institute of Molecular Medicine, John Radcliffe Hospital, Oxford OX3 9DS, United Kingdom; fax: +44-(0) 1865-222-500; E-mail: elouis@worf.molbiol.ox.ac.uk2To whom correspondence should be addressed.
Submitted July 16, 1997.
Recent work has yielded considerable information concerning the structure and function of telomeres and their associated sequences in the budding yeast Saccharomyces cerevisiae. The structure and maintenance of telomeres depends not only on the RNA template and the catalytic subunit of telomerase, but on a number of other proteins. These include proteins involved in assessing DNA damage and cell cycle regulation. There are also non-telomerase mediated processes involved in the normal maintenance of telomeres. In addition to proteins involved in telomere maintenance, there are a number of other proteins involved in the chromatin structure of the region. Many of these proteins have roles in silencing, ageing, segregation and nuclear architecture. The structure of the subtelomeric regions has been well characterised and consists of a mosaic of repeats found in variable copy numbers and locations. Amidst the variable mosaic elements there are small conserved sequences found at all ends that may have functional roles. Recent work shows that the subtelomeric repeats can rescue chromosome ends when telomerase fails, buffer subtelomerically located genes against transcriptional silencing, and protect the genome from deleterious rearrangements due to ectopic recombination. Thus the telomeres of yeast have a variety of roles in the life of the yeast cell beyond the protection of the ends and overcoming the end replication problem associated with linear molecules.
KEY WORDS: telomeres, subtelomeric repeats, telomere associated sequences, telomere position effect, position effect variegation, silencing, ageing, senescence, telomere clustering, nuclear architecture.
Telomeres are important structures for linear eukaryotic chromosomes. For recent discussions of the origin of the telomere concept and the specialised replication problems of chromosome ends, see [1, 2]. In recent years a number of other properties of chromosome ends have been found (see [3-6] for recent reviews). These properties include possible roles in ageing and senescence, transcriptional silencing/chromatin structure, segregation, cell cycle control, chromosome movement, and nuclear architecture. This review will concentrate on recent work dealing with these new properties as well as the mechanisms involved in telomere maintenance.
In addition to the telomere repeats per se, eukaryotic chromosome ends have a mosaic of highly variable repeated sequences adjacent to the telomeres, and at least one taxon, Drosophila [7], has only these repeats with no canonical telomere repeats. Yeast is no exception to this rule, and with the complete genome sequence of S. cerevisiae [8] there is a complete sequence characterisation of the subtelomeric region. A recent comparison among human chromosome ends and all of the yeast ends found a remarkable conservation of overall structure [9]. This conservation is consistent either with common processes leading to the generation and maintenance of common structures, or with there being functional constraints on the structures found in subtelomeric regions. Recent work indicates that some of the subtelomeric region is indeed functional and the potential functions, and consequences of these functions, will be assessed in this review.
The Telomeres of S. cerevisiae
The telomere sequence of S. cerevisiae is a variable repeat of TG1-3. In general there is 300 ± 75 bases of TG1-3 sequence at the ends (see [4] for review). The telomere is maintained by telomerase which consists of an RNA template molecule encoded by TLC1 [10] and a protein complex, the catalytic subunit of which is EST2 [11, 12]. Null mutations in either EST2 or TLC1 result in a senescence phenotype with a loss of several base pairs per division until growth can no longer be maintained [10, 12]. Over-expression of TLC1 results in derepression of telomeric silencing (see below) and shorter telomere tracts. Expression of mutant sequences in the template region of TLC1 causes corresponding changes in the telomere sequence added. A number of other proteins are also involved in telomere maintenance and some of these apparently interact with the EST2. These include EST1 [13] and EST3 [11], both of which show the shortening of telomeres seen in EST2 and TLC1 mutants. CDC13 (EST4) [14-16], an essential gene, has different phenotypes depending on the particular mutation. The cdc13-ts mutants result in the uncontrolled degradation of the C-strand [14] while the cdc13-est mutant results in a phenotype identical to est1, est2, est3 and tlc1 mutants [15].
HDF1 [17], HDF2 [18] (the yeast homologues of Ku70 and Ku80) and TEL1 [19, 20] (a yeast homologue of ATM, the ataxia-telangiectasia gene) are also involved in telomere maintenance; at temperatures less than 30°C they have short telomeres while at 37°C they show the senescence phenotypes of telomerase mutants. The HDF1, and presumably HDF2, gene appears to be acting independently of TEL1, as double mutants are more severely affected than either of the single mutants [17]. A number of other genes and factors appear to be involved in the length of the telomeres per se causing shorter or longer TG1-3 tracts, but not the senescence seen with the above mutations (see [3, 4] for reviews). Some of these are involved in replication and cell-cycle progression such as CDC17 [21, 22] and CDC44 [21].
There is also a cell-cycle controlled degradation of the C-strand, generating G-strand overhangs, that is telomerase independent [23]. This process is likely to be an integral part of the telomere maintenance process. The cell-cycle dependent association of the G-strand overhangs in small artificial linear molecules [24] may reflect interactions of native chromosomal ends.
There appears to be a homeostasis mechanism in place in which telomere length is maintained at an optimum. Rap1p, a DNA-binding protein involved in transcriptional repression and activation, is involved in the measurement of the length and therefore in length control. There is a Rap1p-binding site on average every 20 bp in TG1_3 sequences and rap1 mutations result in changes in the average telomere length. In certain Rap1p mutants there is expansion of the telomere repeat length [25]. Rap1p is one of several telomere-binding proteins that contain a single Myb-repeat called a "telobox" [26]. This expansion of telomere repeats is also seen in S. pombe in some mutants of Taz1p, another Myb-containing protein [27]. The human telomere-binding protein hTrf1p, which also contains a Myb-repeat, has a similar role in telomere length control [28]. The maintenance of the average length of yeast telomeres may be due to a "counting" mechanism of bound Rap1p molecules [29] which is also the case in Kluyveromyces lactis [30]. In addition, homeostasis can be re-established via a rapid recombination mechanism in which large blocks of TG1-3 sequence can be lost in one step at ends above the range of acceptable lengths [31]. This homeostasis appears to be a general property of telomeres and is governed by a telobox-containing protein.
Recombination and Survival in the Absence of Telomerase Activity or a Telomere
When telomere maintenance activities are lost as is the case with tlc1, est2 as well as est1, cdc13-est, est4, hdf1 and hdf2 at 37°C, the cells senesce after many generations as their telomere repeats get shorter. Survivors are recovered with relatively high efficiency and these survivors maintain their chromosome ends by recombination resulting in amplification of tandem arrays of subtelomeric repeats, in particular the Y´ element [32]. This amplification is RAD52 dependent and is likely to involve the internal TG1-3 tracts. In K. lactis a similar phenomenon is observed, with RAD52 dependent recombination among the longer telomere repeats being the cause of survival in cells lacking telomerase RNA. Apparently, one of the properties of the telomere is prevention of high levels of telomeric recombination [33].
Chromatin Structure
The telomere itself is in non-nucleosomal chromatin with a large nuclease protected region of approximately 350 bp called the telosome [34, 35]. The proteins involved in this protected telosome include Rap1p. Others have not been identified but are likely to include the known telomere associated proteins. The telosome is then followed by phased nucleosomes. Gottschling [36] demonstrated that the telomere regions were inaccessible to dam methylase when expressed in yeast, whereas other genomic sequences were methylated, thus implying a different structure to the chromatin. In other organisms the histones in heterochromatin are hypoacetylated and this is also the case for yeast subtelomeric regions [37, 38]. Both the resistance to dam methylase and the hypoacetylation require many of the genes involved in telomere position effect.
Position Effect Variegation or Telomere Position Effect
In artificial structures in which a marker is integrated immediately adjacent to TG1-3 sequences, position effect variegation (PEV), or telomere position effect (TPE), of the marker's expression has been observed [39-42]. This variegated expression is reminiscent of the PEV seen in Drosophila when a gene is placed near heterochromatic regions in chromosomal rearrangements. Telomere position effect variegation is also seen in S. pombe [43]. The suppressed expression state depends on a number of genes including RAP1; SIR2, SIR3 and SIR4 (genes involved in the transcriptional repression of the silent information mating cassettes HML and HMR); NAT1 and ARD1 (protein N-acetyltransferase subunits); HH3 and HH4 (core histones); and is modulated by a number of other factors including RIF1 [44] and RIF2 [45] (Rap1p-interacting factors); and SUM1 [46] (a suppressor of sir mutations) (see reviews [4, 47, 48]). Recent work has added HDA1 [49] and RPD3 [49-51] (members of histone deacetylase complexes), SAS2 [52] (an acetyltransferase homologue) and UBP3 [53] (a SIR4-interacting deubiquitinating enzyme) to the proteins involved in silencing at HML, HMR, and telomeres.
There is clearly a lot of overlap between the silencing seen at telomeres and that seen at HML and HMR. In earlier work concerning TPE no role was found for SIR1, and therefore presumably with the origin recognition complex (ORC), which do have a role in HML and HMR silencing. This is consistent with the fact that the artificial truncations had no autonomously replicating sequences (ARS) nearby. ORC mutations have been identified that are clearly involved with TPE at the artificial structures [54], despite the lack of an ARS, and SIR1 appears to have a role in silencing near native telomere structures which do have an ARS (Pryde and Louis, unpublished). As seen below the subtelomeric structures found at all ends are structurally similar to the HML and HMR E and I sites and these may play a role in regulating the TPE seen at native chromosome ends by involving the ORC and Sir1p proteins as well as accessory factors [47, 48].
Meiotic Recombination near Telomeres
Another phenomenon related to chromatin structure is the meiotically induced double-strand breaks (DSBs) which occur at pre-existing DNase I hypersensitive sites established in vegetative growth [55-57]. These tend to be in promoter regions where nucleosomes are disrupted by proteins bound to enhancer elements, but also occur at other non-promoter sites in the genome. The telomeres and subtelomeric regions have no detectable DSBs [58-60]. This means that the hypersensitive site between the telosome and the phased nucleosomes of the subtelomeric sequences [34, 35] are not recognised by the recombination machinery. The reduced levels of DSBs correlate with the low level of homologous recombination seen in telomeric regions ([61] and Greig, Louis and Borts, unpublished) but are not necessarily consistent with the levels of ectopic recombination seen among telomerically located sequences ([62, 63] and Gorham and Louis, unpublished). This means that ectopic recombination near telomeres is different somehow from homologue recombination and does not require detectable levels of meiotic DSB formation.
Subnuclear Clustering and Localisation of Telomeres and Associated Proteins
Having antibodies to telomere associated proteins has allowed for the localisation of telomeres within the nucleus with maintenance of 3-D structure which in general is not maintained for most fluorescent in situ hybridisation (FISH) protocols. Using anti-Rap1p antibodies it has been found that the telomeres are clustered into a few foci and these foci are near the nuclear periphery [64, 65]. The foci are disrupted by mutations in SIR2, SIR3 and SIR4 [64] and in RLF genes [66]. One of them, RLF2, is part of the chromatin assembly factor [67], the absence of which causes the delocalisation of Rap1p but not of the telomere sequences. Using antibodies to SIR3 and SIR4 in protocols in which immunofluorescent localisation and FISH can be done sequentially, it has now been demonstrated that these proteins colocalise with each other and with the subtelomeric Y´ sequences [68]. Mutations in many genes disrupt the protein foci without disrupting the Y´ foci near the nuclear periphery, though the clusters appear qualitatively "looser" [68].
No mutations have been identified that disrupt the sequence localisation of the telomeres. Only the protein--protein interactions are disrupted. The protein localisation is relevant for silencing and ageing (see next section) but may not be relevant for nuclear architecture and the physical movement of telomeric sequences. A recent study of telomeric sequences on non-replicating plasmids found that the yeast telomeric sequences are necessary and sufficient for blocking DNA rotation in a topoisomerase I assay [69]. The authors propose that the telomeric sequences are anchored via DNA--protein interactions to a nuclear structure. This anchor includes Rap1p but is not dependent on SIR2, SIR3 and SIR4. It appears that the physical locations of the telomeric sequences and the protein foci are separable properties.
Telomeres and Ageing
Telomere length is a good molecular clock for replicative age, as they shorten with cell divisions in somatic tissues of many organisms. Many immortalised cells maintain telomere length by turning on telomerase. These observations have supported the hypothesis (reviewed in [2]) that telomeres may be involved with ageing in some way. In yeast there is also a connection between telomeres and ageing [70, 71] but as will be seen below the connection is not as might have been expected.
A model of the role of telomeres in ageing in which an "AGE" locus near telomeres is transcriptionally repressed due to TPE, with derepression occurring in older cells as the telomere gets shorter, has been proposed for mammalian cell ageing [72]. Indeed, a mutation in one of the genes (SIR4) involved in telomeric silencing causes increased lifespan in yeast [73]. However, this mutation has shorter telomeres [73], not longer, and older yeast cells do not have any decrease in telomere length [74, 75]. Older yeast cells do have a loss of telomeric silencing [75] which is now known to be due to a redistribution of the silencing factors within the nucleus from telomeres to the nucleolus [76]. The rDNA genes may therefore be the "AGE" locus in yeast and the role of telomeres is indirect.
Telomeres and Segregation
Recently, a meiosis specific telomere-binding protein, NDJ1/TAM1, has been identified [77, 78]. Mutations in this gene result in disbursement of meiotic Rap1p foci such that each telomere is unpaired (32 foci increasing to 64), and in an increase in meiosis I non-disjunction and precocious sister segregation events. This increased missegregation is not due to a reduction in reciprocal exchanges but may be due to the loss of cross-over interference which is generally thought to guarantee that all chromosomes get at least one crossover. Distributive segregation, the process by which non-cross-over and non-homologous partners segregate, is also perturbed. It is thought that this gene plays an important role in homologue pairing but not necessarily initiation of pairing.
The Telomere-Associated Sequences or Subtelomeric Region
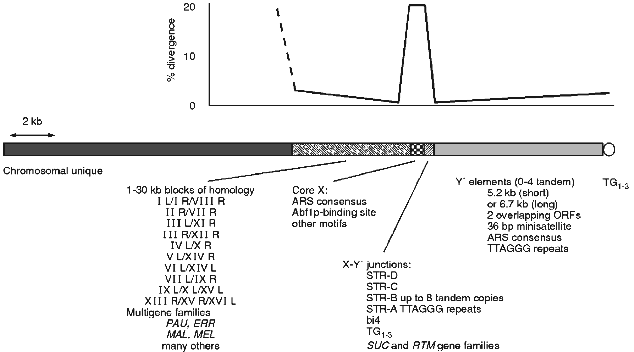
In S. cerevisiae there are a number of repeated sequences found at ends of the chromosomes (see [3, 4] for earlier reviews). Some of these are ORFs, others are non-coding. In general there is a mosaic of different repeats at any given end that varies from end to end and from strain to strain at the same end [3]. This complex mixture and the obviously dynamic nature of the region has made it difficult to assess the common and possible functional components of the subtelomeric region. A picture of a typical chromosome end in yeast can be generated by comparison of all chromosome end sequences (see Fig. 1).
Y´ elements are highly conserved and well characterised yet their origin and function are unknown (see [3] for review). They are found in two major size classes and contain two overlapping ORFs. They also contain an ARS consensus and TTAGGG repeats near the distal end (see Fig. 1). Between X and Y´s are smaller sequences, some combination of which is found at most ends. These subtelomeric repeat, or STR, sequences contain degenerate versions of the vertebrate TTAGGG telomere repeats which are the binding site for the essential gene of unknown function TBF1 [79, 80]. In at least one case one of these STR elements is found in a tandem array of at least 8 identical copies [63]. In two strains there is part of the 4th intron of the mitochondrial encoded cytochrome b [81]. This intron and its sequence context in the STRs is consistent with a transposition into this nuclear location. In both strains there are two copies, one location shared by both and the second distinct for each consistent with a single transposition followed by separate recombinational duplications in the two strains.Fig. 1. The chromosome ends of S. cerevisiae consist of a number of repeated structures that form a mosaic pattern. A generic end is shown here. All ends have a minimal core X element, containing an ARS consensus, in addition to the TG1-3 repeats of the actual telomere. Between the telomere and the core X element there can be up to 4 tandem copies of the highly conserved Y´ element. Between the X and Y´ are usually found short subtelomeric repeats that exist in variable copy number and contain degenerate versions of the vertebrate telomere repeat, TTAGGG. Various other repeated sequences are found in this junction region, including the bi4 intron and the SUC and RTM gene families. On the centromere proximal side of the X are larger less dispersed repeats some as large as 30 kilobases. Within these repeats are the PAU family of ORFs and this region is where the MAL and MEL gene families are found as well as many others. Where there is homology the sequence identity is greatest at the junction between the X and adjacent sequences, dropping off with distance from the X. The X elements are not as homogeneous with an average divergence of 15-20%.
Conserved Elements and Structures
Until recently the mosaic nature has precluded a thorough understanding of the structure of the region. Now that all of the ends of one strain have been sequenced [8], and there is information of ends from other strains and even other closely related species, we can now build a better picture of the structure of the chromosome ends. One can look for and find conserved sequence elements among the mosaic of subtelomeric sequences. The core X element [63, 82], having been continuously refined as more sequences have become available, is now defined as a 475 bp element found in entirety at most ends (29 out of 32 are full length, 31 out of 32 have the ARS- and Abf1p-binding sites) and minimally (at least the ARS consensus) at all ends. Within the full length element in addition to the ARS consensus and Abf1p-binding site there are several other conserved motifs.
The core X element and the adjacent smaller STR elements divide the yeast subtelomeric regions into two domains. The distal domain consists mainly of the tandem arrays of Y´ elements which are highly conserved and found at many ends, but are polymorphic in location between strains, while the proximal domain consists of longer tracts of sequence homology found at only a few ends. It is interesting to note that the sequence identity on either side of the X element is nearly 100% for those ends that share homology but at the X itself the homology is only 85% on average (see Fig. 1). The flanking homology-block identities are reduced with increased distance from the X. This general structure of two domains with different copy number and homology tract-length properties, divided by a small region containing degenerate TTAGGGs and a potential origin of replication, is seen in human telomeres [9]. The conserved structures may indicate a conserved function(s) for a two domain subtelomere region or for having a boundary element.
There is some evidence that the X element does have function. The X element stabilises plasmids containing both a centromere and telomere and mutations in either of the ARS- or Abf1p-binding sites disrupts this stability [83]. However, there is no apparent defect in chromosome stability in the absence of X ([84] and Pryde and Louis, unpublished) but there are other roles it may play. The X element when expressed in the direction towards the telomere causes cell cycle arrest [85]. Although no open reading frame is found in most X elements this indicates some possible role of X RNAs, or the binding of essential factors to X RNAs titrating them away from their normal locations. Consistent with the idea that X expression may be of biological significance is the observation that the telomere region, including the X, is highly expressed upon entry to meiosis [3]. The X element is also a preferred integration target for the Ty5 retrotransposon [86]. This preference for Ty5 integration is not dependent on the actual telomere being close as integrations are efficient at Xs where there is at least one Y´ distal, placing the telomere at least 6 kb away [87]. In the related species S. paradoxus, where Ty5s are active in some strains, the same preference is seen and the X sequences appear to be conserved relative to flanking DNA [87].
X may play additional roles when it comes to the chromatin structure and architecture of the region. When silencing is measured for marker genes inserted in and around the native subtelomeric sequences, very little silencing is found except at the telomere itself and at the X-ARS element (Pryde and Louis, unpublished). The X and its putative binding factors may be regulating the levels of silencing seen. An additional role of X may be in the recombinational barrier between telomeric sequences and the rest of the genome (see below). Telomeric sequences can recombine with each other efficiently resulting in translocational exchanges among telomeres with no detrimental effect. These are sequestered from more interstitial homologous sequences so that detrimental recombination events are suppressed (Huckle and Louis, unpublished). In some cases when X is deleted the barrier is relaxed (Pryde and Louis, unpublished).
Gene Families
There are a number of multigene families that exist either solely or predominantly in the subtelomeric regions (see [3] for review). The best known and most studied have been various genes used in carbon source utilisation such as the SUC, MAL and MEL families. Each family has it's own characteristics. There are a number of other gene families that are less well characterised in terms of origin, function, and population variation.
The SUC gene family consists of several loci, all but one of which, SUC2, are embedded in the subtelomeric region between X and Y´ elements [88, 89]. The SUC2 gene on chromosome IX is found in all strains of S. cerevisiae though in many cases it is a non-functioning allele [90]. The other loci are thought to be spread by ectopic recombination among different chromosome ends [89]. It is interesting to note that those strains with many SUC genes do not have any MEL genes [90], and conversely MEL+ strains do not have SUC genes (other than suc2). This difference may reflect adaptation to different environments from where the strains were isolated (see below). In association with many SUC genes is an new multigene family, RTM, that confers resistance to the toxicity of molasses in many brewing situations [91]. It is found in all brewing strains but very few wine strains [92] again indicating a possible adaptive difference in environment related to the variation. RTM is always found in association with SUC but is not found adjacent to the SUC2 locus.
The MAL gene family consists of 5 loci each with 3 or 4 genes coding for different functions involved in maltose metabolism. Each is subtelomerically located, though in this case all are centromere proximal to the X region [93, 94]. One locus exists as a large tandem array of the region containing the 3 genes [95]. Every strain of S. cerevisiae looked at, as well as those of a close relative S. paradoxus, has the MAL1 locus, though in many cases only one of the 3 genes is functional and the strains are unable to ferment maltose [96]. In experiments where specific constitutive mutations were isolated, the involvement of ectopic interactions with cryptic loci homologous to the MAL genes was discovered [97, 98]. As with the SUC loci, it is thought that recombination among the chromosome ends is involved in the spread and amplification of this gene family.
The MEL gene family is similar to the MAL family in that all copies are subtelomeric and are situated proximal to the X element [99, 100]. In this case though there are many strains with no sequence homology to MEL genes [101-103]. MEL genes are found in related species and several have been cloned and sequenced [104]. The level of divergence of MEL genes between species is consistent with other known sequence divergences; however, it appears that the mutation rate may be different for the MEL genes. The ratio of transitions versus transversions in 3 pairs of homologues between S. cerevisiae and S. paradoxus is about 3.5 to 1 while the ratio in the MEL genes is only 1.7 to 1 indicating either a greater age of divergence or a different rate or type of mutation. The difference could be due to the fact that the MEL genes are a multigene family or that they are subtelomerically located. Sequence comparisons of the subtelomeric regions have lead to the proposal that the region is subject to a different rate or mode of mutation [104].
There are other repeats at telomere regions in yeast. In particular the Ty5 elements and associated LTRs [86]. These retro-elements are reminiscent of the TART and HET-A elements that maintain the chromosome ends of Drosophila [7]. In many organisms there are retro-elements found near telomeres and it is possible that they can serve to maintain chromosome ends in the absence of telomerase [87]. In the case of Drosophila, perhaps, this mechanism was efficient enough that loss of telomerase was not disadvantageous and perhaps was adaptive for the particular selective conditions that Drosophila encounter.
Recombinational and Evolutionary Dynamics
There is clearly a great deal of recombinational exchange occurring among chromosome ends in yeast. These interactions have been measured among the Y´ elements [63, 105]. There is sufficient recombination in vegetative cells to account for the sequence homogenisation; however, the maintenance of two size classes is not predicted by simple models. One possible explanation for this is the fact that the recombination seen is highly non-random which would allow for within size-class homogenisation, and some cross size interactions, but not the complete supplanting of one size class by the other. This non-random interaction is not dependent on the sequence type of the Y´ element but on the particular ends involved. Now that the entire yeast genome is sequenced it is clear that the non-random choice of recombinational partners is completely correlated with shared sequence homologies proximal to the X element.
The sequence comparisons on both sides of X, as mentioned above, and the sequences flanking the SUC, MAL and MEL multigene families, indicate that the recombinational dynamics of the regions are not simple and may involve some illegitimate interactions or recombination between very short tracts of homology. X appears to be a boundary element for homogenisation processes which occur on either side. There also appears to be a recombinational boundary between telomerically located sequences and the rest of the genome (see above) which involves many genes (Timbrell, Huckle, Underwood, Gorham, Borts and Louis, in preparation) but not the SIR genes.
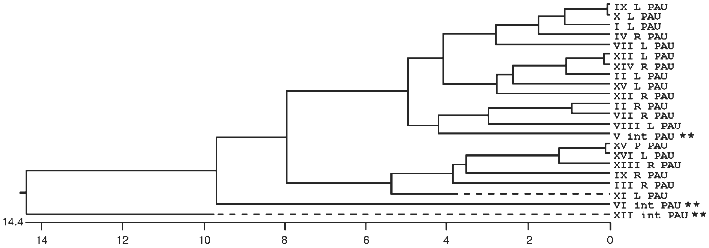
In many organisms the subtelomeric regions are used to generate variation in genes used for adaptive purposes. This is most evident in the parasites and pathogens that have to evade host immune systems. We now see evidence in S. cerevisiae that the region is adaptive for different environments (MEL versus SUC for example) and that genetic variation is generated by recombination mechanisms analogous to that seen in Trypanosomes and Plasmodium (the MAL genes). The ability of yeast to sequester the telomeric regions from the rest of the genome allows recombination among dispersed sequences in one domain without detrimental effect, while the rest of the genome is not susceptible to such recombination. Evidence of this sequestering into two domains can be seen in the comparison of sequences of members of multigene families, such as the PAU family [106], where some members are interstitially located and others are subtelomeric (see Fig. 2).
Overall Model of the Nucleus and the Architecture of the TelomeresFig. 2. A phylogenetic tree of relatedness using the coding sequence plus 75 base pairs of flanking sequence for all 22 members of the PAU family. The tree was built with the MegalignTM software from DNAstar and uses the Clustal method. There are three interstitial members (**) of the family and two of them are the most divergent as seen in the diagram. When longer flanking sequences are also included in the analysis, the third interstitial copy become the third most divergent member. This sequence relatedness is consistent with subtelomeric homogenisation while the interstitial copies are evolving independently without any interaction with each other or the subtelomeric copies.
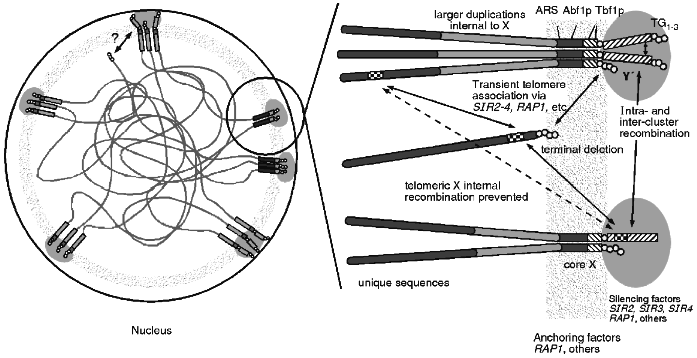
The combined data of telomere clustering, telomeric protein foci, nucleolar interactions and distribution of silencing factors, and recombinational interactions leads to a unified view of the subnuclear organisation of telomeres (see Fig. 3). This physical model can explain the recombinational interactions seen, including the non-random choice of partners, the apparent barrier to homogenisation across X and the apparent sequestering of telomere regions from the rest of the genome. A nuclear structure involving proteins bound to the telomere as well as to telomere associated sequences like X may form a physically rigid structure that results in the observed recombinational phenomenon. It may also explain the lack of meiotic DSBs as the sequences in the region are bound up in this structure and not accessible to the nuclease.
This structure can also explain the differences between TPE seen in artificially truncated telomeres and at native telomeres. The artificial truncation may only be transiently associated via protein--protein interactions with other telomeres while a native telomere is in the nuclear structure imposing the architecture illustrated. Perhaps the "OFF" state is when the truncated telomere is at a cluster while in the "ON" state the truncated telomere is not associated with a cluster.Fig. 3. One view of the architecture of the nucleus. The telomeres appear to be clustered and near the nuclear periphery. These clusters of sequences colocalise with foci of proteins involved in silencing which can be redistributed within the nucleus during ageing or in particular mutations. The anchoring of the telomere clusters to some nuclear structure appears to be independent of the silencing factors and may involve other proteins. This anchored architecture may explain several properties of telomere regions in yeast. Silencing at telomeres is more tightly regulated than silencing on terminally truncated telomeres and this may be a reflection of the transient association of the truncated ends. Recombination is restricted for homologues but not for ectopically located sequences which may be the result of the anchoring. Recombination is repressed between the distal sequences and interstitial sequences which may not be able to freely move across the architectural structure.
There are clearly a number of properties of telomeres that are important to the cell that go well beyond the protection of the end from degradation or fusions. The near future looks promising for the further elucidation of these properties and their underlying mechanisms. An important consideration is whether the lessons learned from yeast apply to life in general. The shared homologies in genes as well as structures between yeast and many other organisms indicate that these lessons will be generally applicable, which has important implications for models of heterochromatin, nuclear architecture, and ageing.
We would like to thank Rhona Borts and Hazel Gorham for comments and criticisms of this manuscript. We would also like to thank Raymond Wellinger, Daniel Voytas, Michael Dresser, Shirleen Roeder, Matti Korhola, and Michel Aigle for sending preprints and proofs of work prior to publication, and members of the lab for use of unpublished results. The authors are supported by The Wellcome Trust and in part by the EU yeast genome projects.
LITERATURE CITED
1.Gall, J. G. (1995) in Telomeres (Blackburn,
E. H., and Greider, C. W., eds.) Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, New York, pp. 1-10.
2.Olovnikov, A. M. (1996) Exp. Gerontol.,
31, 443-448.
3.Louis, E. J. (1995) Yeast, 11,
1553-1573.
4.Zakian, V. A. (1996) Ann. Rev. Genet.,
30, 141-172.
5.Wellinger, R. J., and Sen, D. (1997) Eur. J.
Cancer, in press.
6.Pardue, M. L. (1994) Curr. Opin. Gene Dev.,
4, 845-850.
7.Mason, J. M., and Biessmann, H. (1995) Trends
Genet., 11, 58-62.
8.Goffeau, A., et al. (1997) Nature,
387, 1-105.
9.Flint, J., Bates, G. P., Clark, K., Dorman, A.,
Willingham, D., Roe, B. A., Micklem, G., Higgs, D. R., and Louis, E. J.
(1997) Human Mol. Genet., in press.
10.Singer, M. S., and Gottschling, D. E. (1994)
Science, 266, 404-409.
11.Lendvay, T. S., Morris, D. K., Sah, J.,
Balasubramanian, B., and Lundblad, V. (1996) Genetics,
144, 1399-1412.
12.Lingner, J., Hughes, T. R., Shevchenko, A., Mann,
M., Lundblad, V., and Cech, T. R. (1997) Science, 276,
561-567.
13.Lundblad, V., and Szostak, J. W. (1989)
Cell, 57, 633-643.
14.Garvik, B., Carson, M., and Hartwell, L. (1995)
Mol. Cell Biol., 15, 6128-6138.
15.Nugent, C. I., Hughes, T. R., Lue, N. F., and
Lundblad, V. (1996) Science, 274, 249-252.
16.Lin, J.-J., and Zakian, V. A. (1996) Proc.
Natl. Acad. Sci. USA, 93, 13760-13765.
17.Porter, S. E., Greenwell, P. W., Ritchie, K. B.,
and Petes, T. D. (1996) Nucleic Acids Res., 24,
582-585.
18.Boulton, S. J., and Jackson, S. P. (1996)
Nucleic Acids Res., 24, 4639-4648.
19.Morrow, D. M., Tagle, D. A., Shiloh, Y., Collins,
F. S., and Hieter, P. (1995) Cell, 82, 831-840.
20.Greenwell, P. W., Kronmal, S. L., Porter, S. E.,
Gassenhuber, J., Obermaier, B., and Petes, T. D. (1995) Cell,
82, 823-829.
21.Adams, A. K., and Holm, C. (1996) Mol. Cell
Biol., 16, 4614-4620.
22.Carson, M. J., and Hartwell, L. (1985)
Cell, 42, 249-257.
23.Dionne, I., and Wellinger, R. J. (1996) Proc.
Natl. Acad. Sci. USA, 93, 13902-13907.
24.Wellinger, R. J., Ethier, K., Labrecque, P., and
Zakian, V. A. (1996) Cell, 85, 423-433.
25.Kyrion, G., Boakye, K. A., and Lustig, A. J.
(1992) Mol. Cell Biol., 12, 5159-5173.
26.Bilaud, T., Koering, C. E., Binet-Brasselet, E.,
Ancelin, K., Pollice, A., Gasser, S. M., and Gilson, E. (1996)
Nucleic Acids Res., 24, 1294-1303.
27.Cooper, J. P., Nimmo, E. R., Allshire, R. C., and
Cech, T. R. (1997) Nature, 385, 744-747.
28.Van Steensel, B., and de Lange, T. (1997)
Nature, 385, 740-743.
29.Marcand, S., Gilson, E., and Shore, D. (1997)
Science, 275, 986-990.
30.Krauskopf, A., and Blackburn, E. H. (1996)
Nature, 383, 354-357.
31.Li, B. B., and Lustig, A. J. (1996) Genes
Dev., 10, 1310-1326.
32.Lundblad, V., and Blackburn, E. H. (1993)
Cell, 73, 347-360.
33.McEachern, M. J., and Blackburn, E. H. (1996)
Genes Dev., 10, 1822-1834.
34.Wright, J. H., Gottschling, D. E., and Zakian, V.
A. (1992) Genes Dev., 6, 197-210.
35.Wright, J. H., and Zakian, V. A. (1995)
Nucleic Acids Res., 23, 1454-1460.
36.Gottschling, D. E. (1992) Proc. Natl. Acad.
Sci. USA, 89, 4062-4065.
37.Braunstein, M., Rose, A. B., Holmes, S. G.,
Allis, C. D., and Broach, J. R. (1993) Genes Dev., 7,
592-604.
38.Braunstein, M., Sobel, R. E., Allis, C. D.,
Tuner, B. M., and Broach, J. R. (1996) Mol. Cell Biol.,
16, 4349-4356.
39.Gottschling, D. E., Aparicio, O. M., Billington,
B. L., and Zakian, V. A. (1990) Cell, 63, 751-762.
40.Aparicio, O. M., Billington, B. L., and
Gottschling, D. E. (1991) Cell, 66, 1279-1287.
41.Renauld, H., Aparicio, O. M., Zierath, P. D.,
Billington, B. L., Chhablani, S. K., and Gottschling, D. E. (1993)
Genes Dev., 7, 1133-1145.
42.Aparicio, O. M., and Gottschling, D. E. (1994)
Genes Dev., 8, 1133-1146.
43.Nimmo, E. R., Cranston, G., and Allshire, R. C.
(1994) EMBO J., 13, 3801-3811.
44.Hardy, C. F., Sussel, L., and Shore, D. (1992)
Genes Dev., 6, 801-814.
45.Wotton, D., and Shore, D. (1997) Genes
Dev., 11, 748-760.
46.Chi, M. H., and Shore, D. (1996) Mol. Cell
Biol., 16, 4281-4294.
47.Shore, D. (1995) in Telomeres (Blackburn,
E. H., and Greider, C. W., eds.) Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, pp. 139-192.
48.Palladino, F., and Gasser, S. M. (1994) Curr.
Opin. Cell. Biol., 6, 373-379.
49.Rundlett, S. E., Carmen, A. A., Kobayashi, R.,
Bavykin, S., Turner, B. M., and Grunstein, M. (1996) Proc. Natl.
Acad. Sci. USA, 93, 14503-14508.
50.DeRubertis, F., Kadosh, D., Henchoz, S., Pauli,
D., Reuter, G., Struhl, K., and Spierer, P. (1996) Nature,
384, 589-591.
51.Vannier, D., Balderes, D., and Shore, D. (1996)
Genetics, 144, 1343-1353.
52.Ehrenhofer-Murray, A. E., Rivier, D. H., and
Rine, J. (1997) Genetics, 145, 923-934.
53.Moazed, D., and Johnson, A. D. (1996)
Cell, 86, 667-677.
54.Fox, A. F., Ehrenhofer-Murray, A. E., Loo, S.,
and Rine, J. (1997) Science, 276, 1547-1551.
55.Wu, T. C., and Lichten, M. (1995)
Genetics, 140, 55-66.
56.Ohta, K., Shibata, T., and Nicolas, A. (1994)
EMBO J., 13, 5754-5763.
57.Wu, T. C., and Lichten, M. (1994) Science,
263, 515-518.
58.Klein, S., Zenvirth, D., Sherman, A., Ried, K.,
Rappold, G., and Simchen, G. (1996) Nature Genet., 13,
481-484.
59.Zenvirth, D., Arbel, T., Sherman, A., Goldway,
M., Klein, S., and Simchen, G. (1992) EMBO J., 11,
3441-3447.
60.Louis, E. J. (1997) in Methods in
Microbiology: Yeast Gene Analysis (Tuite, M. F., and Brown, A. J.
P., eds.) Academic Press, in press.
61.De Steensma, H., de Jonge, P., Kaptein, A., and
Kaback, D. B. (1989) Curr. Genet., 16, 131-137.
62.Horowitz, H., Thorburn, P., and Haber, J. E.
(1984) Mol. Cell. Biol., 4, 2509-2517.
63.Louis, E. J., Naumova, E. S., Lee, A., Naumov,
G., and Haber, J. E. (1994) Genetics, 136, 789-802.
64.Palladino, F., Laroche, T., Gilson, E., Axelrod,
A., Pillus, L., and Gasser, S. M. (1993) Cell, 75,
543-555.
65.Klein, F., Laroche, T., Cardenas, M. E., Hofmann,
J. F., Schweizer, D., and Gasser, S. M. (1992) J. Cell. Biol.,
117, 935-948.
66.Konkel, L. M. C., Enomoto, S., Chamberlain, E.
M., Mccunezierath, P., Iyadurai, S. J. P., and Berman, J. (1995)
Proc. Natl. Acad. Sci. USA, 92, 5558-5562.
67.Enomoto, S., McCune-Zierath, P. D., Gerami-Nejad,
M., Sanders, M. A., and Berman, J. (1997) Genes Dev.,
11, 358-370.
68.Gotta, M., Laroche, T., Formenton, A., Maillet,
L., Scherthan, H., and Gasser, S. M. (1996) J. Cell Biol.,
134, 1349-1363.
69.Mirabella, A., and Gartenberg, M. R. (1997)
EMBO J., 16, 523-533.
70.Kennedy, B. K., and Guarente, L. (1996) Trends
Genet., 12, 355-359.
71.Shore, D. (1995) Curr. Biol., 5,
822-825.
72.Wright, W. E., and Shay, J. W. (1992) Trends
Genet., 8, 193-197.
73.Kennedy, B. K., Austriaco, N. R., Jr., Zhang, J.,
and Guarente, L. (1995) Cell, 80, 485-496.
74.D'Mello, N. P., and Jazwinski, S. M. (1991) J.
Bacteriol., 173, 6709-6713.
75.Smeal, T., Claus, J., Kennedy, B., Cole, F., and
Guarente, L. (1996) Cell, 84, 633-642.
76.Kennedy, B. K., Gotta, M., Sinclair, D. A.,
Mills, K., McNabb, D. S., Murthy, M., Pak, S. M., Laroche, T., Gasser,
S. M., and Guarente, L. (1997) Cell, 89, 381-391.
77.Chua, P. R., and Roeder, G. S. (1997) Genes
Dev., in press.
78.Conrad, M. N., Dominguez, A. M., and Dresser, M.
E. (1997) Science, in press.
79.Brigati, C., Kurtz, S., Balderes, D., Vidali, G.,
and Shore, D. (1993) Mol. Cell. Biol., 13,
1306-1314.
80.Liu, Z., and Tye, B. (1991) Genes Dev.,
5, 49-59.
81.Louis, E. J., and Haber, J. E. (1991) Curr.
Genet., 20, 411-415.
82.Pryde, F. E., Huckle, T. C., and Louis, E. J.
(1995) Yeast, 11, 371-382.
83.Enomoto, S., Longtine, M. S., and Berman, J.
(1994) Genetics, 136, 757-767.
84.Murray, A. W., and Szostak, J. W. (1986) Mol.
Cell. Biol., 6, 3166-3172.
85.Akada, R., Yamamoto, J., and Yamashita, I. (1997)
Mol. Gen. Genet., 254, 267-274.
86.Zou, S., Wright, D. A., and Voytas, D. F. (1995)
Proc. Natl. Acad. Sci. USA, 92, 920-924.
87.Zou, S., Kim, J. M., and Voytas, D. F. (1996)
Nucleic Acids Res., 24, 4825-4831.
88.Carlson, M., and Botstein, D. (1983) Mol.
Cell. Biol., 3, 351-359.
89.Carlson, M., Celenza, J. L., and Eng, F. J.
(1985) Mol. Cell. Biol., 5, 2894-2902.
90.Naumov, G. I., Naumova, E. S., Sancho, E. D., and
Korhola, M. P. (1996) FEMS Microbiol. Lett., 135,
31-35.
91.Ness, F., and Aigle, M. (1995) Genetics,
140, 945-956.
92.Denayrolles, M., Pinto de Villechenon, E.,
Lonvaud-Funel, A., and Aigle, M. (1997) Curr. Genet., in
press.
93.Charron, M. J., and Michels, C. A. (1988)
Genetics, 120, 83-93.
94.Charron, M. J., Read, E., Haut, S. R., and
Michels, C. A. (1989) Genetics, 122, 307-316.
95.Michels, C. A., Read, E., Nat, K., and Charron,
M. J. (1992) Yeast, 8, 655-665.
96.Naumov, G. I., Naumova, E. S., and Michels, C. A.
(1994) Genetics, 136, 803-812.
97.Gibson, A. W., Wojciechowicz, L. A., Danzi, S.,
Zhang, B., Kim, J. H., Hu, Z., and Michels, C. A. (1997)
Genetics, in press.
98.Wang, J. F., and Needleman, R. (1996)
Genetics, 142, 51-63.
99.Naumov, G., Turakainen, H., Naumova, E., Aho, S.,
and Korhola, M. (1990) Mol. Gen. Genet., 224,
119-128.
100.Turakainen, H., Naumov, G., Naumova, E., and
Korhola, M. (1993) Curr. Genet., 24, 461-464.
101.Turakainen, H., Aho, S., and Korhola, M. (1993)
Appl. Environ. Microbiol., 59, 2622-2630.
102.Naumov, G. I., Naumova, E. S., and Korhola, M.
P. (1995) FEMS Microbiol. Lett., 127, 41-45.
103.Naumov, G. I., Naumova, E. S., Turakainen, H.,
and Korhola, M. (1996) Genet. Res. Camb., 67,
101-108.
104.Naumova, E. S., Turakainen, H., Naumov, G. I.,
and Korhola, M. (1996) Mol. Gen. Genet., 253,
111-117.
105.Louis, E. J., and Haber, J. E. (1990)
Genetics, 124, 547-559.
106.Viswanathan, M., Muthukumar, G., Cong, Y. S.,
and Lenard, J. (1994) Gene, 148, 149-153.