DNA Reorganization and Biological Aging. A Review
H. D. Osiewacz1,2 and A. Hamann1
1Botanisches Institut, Molekulare Entwicklungsbiologie und Biotechnologie, Johann Wolfgang Goethe-Universität, Marie-Curie-Str. 9, D-60439 Frankfurt/Main, Germany; E-mail: Osiewacz@em.uni-frankfurt.de2To whom correspondence should be addressed.
Submitted July 2, 1997.
Biological aging is a fundamental process observed in almost all living beings. It is characterized by a progressive impairment of biological systems leading to an increase in age-related mortality. The understanding of the mechanisms of aging is of particular interest not only from a general point of view, but it has wide implications for both the human individual and society. Specifically, understanding the basic mechanisms leading to human aging will certainly provide novel strategies to deal with the many different and severe age-related diseases (e.g., different types of cancer, Alzheimer's disease, cardiovascular diseases) and can be expected to increase the quality of life in old age significantly. Despite this obvious significance, it is amazing how little is known about the basis of aging. Currently, it is not clear whether there exists a single conserved mechanism by which all biological systems age, nor is it clear whether or not such a mechanism accounts for aging in a single species. However, beside these and many other open issues, it is clear today that there is a genetic component of aging. It is also clear that age-dependent changes of genetic information play an important role. The processes leading to the observed age-related accumulation of DNA alterations is dependent on two major factors: the rate at which the alterations occur and the ability of the biological system to repair them. In addition, there are a number of different types of DNA alterations known. One group of changes are unspecific and occur randomly. Other alterations result from the differential activity of specific genetic traits (e.g., age-related genes, mobile genetic elements). Moreover, DNA changes may be subtle, occurring at the nucleotide level, or may include larger regions of the genetic information. In this review we focus on the latter type of age-related DNA reorganizations. We will refer to reorganizations which are of special relevance in different biological systems.
KEY WORDS: aging, DNA reorganization, mitochondrial DNA, age-related genes, reactive oxygen species.
REORGANIZATION OF THE NUCLEAR GENOME
Chromosomal Aberrations
Among the gross reorganizations of the nuclear genome which are known to occur in different biological systems, an age-dependent increase in anomalies in chromosome numbers, aneuploidy, has been repeatedly reported (for review, see [1]). In cultured lymphocytes the percentage of aneuploid nuclei increases with the age of the donor. In some studies, chromosome losses were found to be much more frequent than the gain of chromosomes, suggesting a selective advantage of monosomic or a disadvantage of trisomic cells. Alternatively, it was suggested that chromosome loss is more abundant due to an age-dependent increase of physical abnormalities of the mitotic spindle apparatus [2, 3]. Interestingly, the X chromosome was found to be more frequently lost than other chromosomes. More specifically, the preferential loss of the heterochromatic, inactive X chromosome was striking in one study [2]. In contrast to somatic cells, a gain of chromosomes was found to occur with a significantly elevated incidence in germline cells of older men [4]. Also in this study, in which 400,000 sperm cells from 24 individuals of ages between 18 to 60 years were analyzed, the sex chromosomes were found to occur in an abnormal number leading to XY, YY, and XX disomic nuclei.
Apart from changes in chromosome numbers, structural abnormalities of the chromosomes were reported to increase during aging. Martin et al. [5] found a sixfold increase of chromosomal aberrations in kidney cells from old mice compared to young mice. Basically the same results were obtained with cultured lymphocytes from human donors of different ages [6]. In contrast, Bender et al. [7] analyzed a large number of lymphocytes from about 500 individuals and could not detect an age-related change in the mean frequency of chromosome aberrations. Interestingly, somatic cells from patients with Werner's syndrome, a model of accelerated aging, were found to display an elevated incidence of translocations, deletions, or inversions. These data suggest that an increased somatic mutation rate plays an important role in this syndrome [8].
Beside these general chromosomal changes, reorganizations of the rDNA region, a region of the nuclear genome consisting of repeated rRNA genes, was emphasized to represent a region in which specific age-related changes in chromosome structure occur [9]. However, the data derived from different systems (e.g., different tissues of mice, beagle dogs, and humans) are conflicting and may suggest that this type of chromosomal reorganization may be tissue and species specific [10].
Telomere Shortening and Replicative Senescence
Somatic cells of vertebrates show only a limited capacity to proliferate. This phenomenon, called replicative senescence, is controlled by a molecular counting mechanism (molecular clock) which was suggested to be related to a specific type of DNA reorganization occurring at the ends of the chromosomes, the telomeres [11]. During proliferation of cells, the chromosomes are replicated by the conventional DNA polymerase, an enzyme which is dependent on a RNA primer to initiate replication. After removal of the primer, the newly replicated DNA strand is shortened at the 5´-end. This process is repeated every cell cycle and thought to be responsible for the observed time-dependent reduction in proliferation ability. In the past, some striking data supporting the role of telomere shortening in replicative senescence have been reported. First, Harley et al. [12] demonstrated a progressive shortening of telomeres in human fibroblast cell cultures and in other somatic cell types. Second, immortal tumor cells possess an active ribonucleoprotein, called telomerase. Telomerase appears to be important in stabilizing chromosome ends [13]. Third, Harley et al. [14] reported that telomeres in germ line cells are longer than in somatic cells. In germline cells, telomeres remain stable during aging of the donor. These data led to the proposal of the telomere theory of aging and immortalization [14]. According to this theory maintenance of the telomeres in germline cells is due to the activity of telomerase, a ribonucleoprotein which extends a DNA strand at the 3´-end in the absence of a chromosomal template. During differentiation, the enzyme becomes repressed and the telomeres therefore progressively shorten. At a critical stage, when the "Hayflick limit" was exhausted, at least one chromosome has lost a substantial part of its telomeres and the cell stops dividing. Mutation or expression of viral oncogenes allows some cells to overcome the Hayflick limit. While dividing, the telomeres of these cells shorten until those of most chromosomes reach a critical minimum length of about 1.5 kbp [13]. At this stage, termed "crisis", most cells die. Only a few cells may escape "crisis" by further mutations evoking telomerase activity and a stabilization of the chromosome ends. Although this model conclusively explains replicative senescence occurring in different cell types, some recent data were reported which either suggest a modification of the model or which are not consistent with a general role of telomere shortening in respect to aging of all biological systems.
According to the model, tumor cells arise after telomeres were shortened in an early stage of tumorigenesis and after a subsequent activation of telomerase. In contrast to this view, Broccoli et al. [15] could not detect a significant shortening of telomeres in mouse mammary tumors although telomerase activity and telomerase RNA was upregulated. The expression of the telomerase RNA component in mouse mammary tumor cells therefore appears not to be linked to a global loss of telomere function as a result of telomere shortening, but rather is correlated with cell proliferation. Further contradictory data were reported for a plant system. In barley, Kilian et al. [16] found that telomere length decreased in vivo during embryo and inflorescence development. However, during undifferentiated growth of cell cultures derived from immature embryos, the telomeres extended up to three times the length of those of the donor tissue. This increase occurs soon after tissue was explanted from immature embryos (callus initiation). Moreover, callus cultures derived from leafs showed a dramatic increase in telomere length (up to sixfold) within six months of cultivation. These findings are in contrast to what has been found in human cell clones where telomeres shorten until the culture reaches crisis. They may be due to a very early activation of telomerase during the initiation of cell cultures. Interestingly, all investigated long-term callus cultures had lost their potential to regenerate a whole plant. This is perhaps a sign of a general inconsistency of telomere elongation and differentiation.
While the above mentioned recent findings can be explained by a modification of the original telomere theory, investigations reported for two fungal aging models are contradictory to the suggestion that telomere shortening is a general and evolutionary conserved cause of aging. In baker's yeast Saccharomyces cerevisiae it was found that the telomere length is kept within a narrow size range throughout lifespan and seems not to vary with environmental conditions [17-19]. Also, in the filamentous ascomycete Podospora anserina no significant telomere shortening was observed during aging [20]. The lack of telomere shortening in these lower organisms is not surprising because they markedly differ in their general organization from higher systems. In both fungi, a clear distinction between cells of the germ line and somatic cells is not possible. In yeast, a unicellular organism, the single diploid cell resulting from the fusion of two haploid parent cells becomes converted to four ascospores. After karyogamy, the parents of a sexual cross are no longer existing as individuals. On the other hand, the filamentous fungus Podospora anserina represents a system at the border between unicellular and true multicellular organisms. Although the mycelium as the vegetation body is separated by cross walls, a cytoplasmic continuity between the different compartments exists due to a central pore in every cross wall, the so-called septum. This group of filamentous fungi therefore rather represent coenocytes than real multicellular organisms. In addition, the ability to form sexual organs is not restricted to certain parts of the vegetation body and not to a particular time during lifespan.
Transposable Elements
One source of genetic elements which are known to lead to gross DNA rearrangements are the different transposable elements found in almost all eukaryotes. These elements were suggested to be involved in aging [21]. Unfortunately, until now, neither this hypothesis in general nor the exact mechanism by which they may lead to aging has been proved by solid data. However, recently an interesting hypothesis was put forward which explains the relevance of transposable elements with respect to aging by the induction of the mobility of these elements by various stress conditions [22]. Once activated by genomic stress (e.g., DNA damage), the elements may cause further DNA reorganizations (for review see [23]) leading to an accumulation of DNA reorganizations. In the next paragraphs a few examples are mentioned which, although not yet tightly linked to aging, support at least certain aspects of this general idea.
1. Saccharomyces cerevisiae. In search of genes which are differentially expressed under stress conditions, several so-called DDR genes (DNA damage responsive genes) were isolated which were found to be members of the Ty retrotransposons family [24, 25]. Transposition was shown to be triggered by DNA damage [25]. After UV irradiation, Ty transposition to the ADH2 and ADH4 loci, respectively, occurred up to 11-fold and 17-fold more frequently. However, although this is a good example relating the mobility of a mobile element to genomic stress a relationship to aging of S. cerevisiae remains to be demonstrated.
2. Caenorhabditis elegans. In Caenorhabditis elegans, an increase of somatic excision of the transposable element Tc1 was observed during aging [26]. The excision rate at the analyzed Tc1 locus increased from approximately 1% in first-stage larvae to about 3-4% in four-day-old worms. In adult worms, the maximum excision rate reaches about 17%. Since adult worms are consisting of a well defined number of non-dividing cells these data suggest that transposition of this mobile element is not dependent on DNA replication.
3. Drosophila. In Drosophila, Levis et al. [27] first isolated and characterized the terminal end of an intact telomere. The Drosophila telomeres lack short tandem repeats but are composed of several tandemly arranged elements of two families. One type of element is the non-long terminal repeat retrotransposon, TART (telomere-associated retrotransposon) and the other one is HeT-A. The HeT-A element has previously been shown to transpose to broken chromosome ends [28, 29]. These telomere-specific elements have no ability to prevent terminal loss but possess a mechanism to expand [27]. They are never found to disrupt genes and transpose only to the ends of chromosomes.
In contrast to these unusual elements involved in chromosome stabilization, other transposable elements of Drosophila are known which destabilize the nuclear genome. They lead to an increased loss of replication ability either via transposition or via recombination (for review see [22]). Transposition of one member of this type of transposable elements, the P element, was found to negatively effect lifespan of male flies with different numbers of P elements in their genome [30]. Subsequently, it was shown that just one somatically active P element is sufficient to shorten lifespan [31].
4. Mammals. Short interspersed elements (SINEs) and long interspersed repeat elements (LINEs) are highly repetitive components of the mammalian genome. SINEs possess several features of retrotransposable elements. For example, the primate Alu family is highly transcribed and includes an internal RNA polymerase III promoter [32]. Although both types of the elements are potentially able to transpose, no evidence has been reported for a relationship between age-related DNA instabilities and SINE transposition. However, in immortal rat chloroleukemia cells LINE activity was found to correlate with cell death [33]. The increased numbers of L1 copies (about 300,000 per cell) integrate into the nuclear DNA, thereby inducing lethal mutations and cell death. Cellular death was found to be inducible by UV light or ionizing radiation via the activation of this transposable element [34].
REORGANIZATION OF THE MITOCHONDRIAL GENOME
Mitochondrial Dysfunction in Filamentous Fungi
1. Circular Plasmids. The first clear evidence demonstrating the role of extrachromosomal genetic traits in the control of lifespan was derived from a detailed analysis of senescence in the filamentous fungus Podospora anserina (for review see [35-38]). In this simple eukaryotic organism aging is defined by a time-dependent change of the characteristics of a growing culture including an increase in pigmentation, a decreased formation of aerial hyphae, and a decrease in the growth rate. Finally, the culture stops growing and dies at the hyphal tips [39]. Genetic and molecular analyses of different wild-type strains and of various mutants revealed that lifespan is controlled by nuclear and extranuclear genetic traits [40] (for review see [41]). The latter type of factors were demonstrated to be the mitochondrial DNA (mtDNA). The mtDNA of P. anserina and of a few other fungi is extremely unstable. During aging, certain parts become amplified as circular, autonomous elements (circular plasmids). The element most often found in senescent cultures, plDNA or alphasenDNA, is a derivative of the first intron of the cytochrome c oxidase I gene subunit I (COI) [42-45]. The activity of this "mobile intron" appears to be related to the gross mtDNA reorganizations which occur as Podospora cultures age [46, 47]. As a consequence, energy production is compromised leading to death of senescent cultures of this obligate aerobe.
Investigations of about the last five years revealed some insight into the molecular mechanisms leading to the observed age-related mtDNA instabilities. Specifically, it was found that the pl-intron is able to transpose from its resident position in the COI gene to another, ectopic position. This process appears to proceed via an RNA intermediate (retrotransposition) and results in a duplication of the intron sequence in the same mtDNA molecule. These sequences are prone to recombination processes leading to defective mtDNAs [48]. Interestingly, a reverse transcriptase encoded by a long open reading frame of the pl-intron appears to be involved in the mobility of the intron [49]. In addition to the control by a mitochondrially encoded factor, nuclear factors may be involved in the transposition process and the subsequent age-related mtDNA reorganizations. The analyses of certain nuclear long-lived mutants may provide clues towards the identification and characterization of this type of factors involved in maintaining the integrity of the mtDNA of P. anserina which, without any doubt, is relevant for aging in this eukaryotic aging model and also in other systems. Finally, other mtDNA regions which give rise to the formation of other age-related amplification products, e.g., ßsenDNA, gammasenDNA [50, 51], have to be incorporated into the detailed mechanism leading to the formation of defective mtDNA molecules and to mitochondrial dysfunction.
Extensive mtDNA reorganizations related to the activity of circular plasmids have not only been found to be of significance in P. anserina but also in certain strains of two species of the genus Neurospora. In Neurospora crassa and N. intermedia a number of growth-impaired mutants were selected from two wild-type strains (for review see [52]). In these two strains the circular plasmids, Mauriceville and Varkud, were identified. Growth of strains at elevated temperatures (37 instead of 25°C) resulted in altered mtDNAs which become suppressive and accumulated leading to mitochondrial dysfunction and senescence [53].
2. Linear Plasmids in Fungi. In addition to circular plasmids a few linear plasmids, belonging to the group of invertrons, were found to play a major role in the control of lifespan. In certain Neurospora crassa and N. intermedia strains derived from the islands of Hawaii, senescence is initiated by the integration of this type of element into the mitochondrial genome [54-56]. In juvenile cultures, these plasmids are autonomously self-replicating elements. After integration of the plasmids into both coding and noncoding regions, the defective mtDNA molecules become suppressive to the wild-type mtDNA. The mechanism leading to suppressivity is still not understood but appears to be of major interest to understand aging in these species. Here nuclear genes may also play a major role.
In contrast to limiting lifespan, a twelvefold increase in lifespan was found in an extrachromosomal longevity mutant of P. anserina to be related toa linear plasmid, pAL2-1. Lifespan increase was dependent on the ability of the plasmid to integrate into the mtDNA. Strikingly, the linear element interferes with the pl-intron leading to a decreased amplification of this circular, autonomous element [57-60]. Unfortunately, the molecular details of this interference are not understood.
3. Ultra-Short Invasive Elements in Podospora. In P. anserina another type of nucleotide sequence, the undecamer GGCGCAAGCTC, may be involved in the generation of gross mtDNA reorganizations. Beside two other so-called mitochondrial ultra-short invasive elements (MUSEs) this sequence is present in 33 copies in the mtDNA, an unexpectedly high number. Some MUSE were found in close proximity to the boundaries of major rearrangement sites in the mtDNA [61]. Moreover, Jamet-Vierny et al. [62] identified two MUSE sequences flanking the junction sites of betasenDNA. It therefore is reasonable to assume that MUSEs play a role in recombination events and senescence. Several hints indicate that these elements are mobile via a target DNA-primed reverse transcription step and the activity of a reverse transcriptase [61]. However, neither the origin of the MUSE RNA nor the definitive role of these sequences in the control of aging is clear.
Mitochondrial Dysfunction in Mammals
In contrast to the almost quantitative age-related accumulation of defective mtDNA molecules in filamentous fungi, mtDNAs of mammals appear to be quite stable. Nevertheless, a number of maternally inherited human diseases are known which result from mtDNA mutations (for review see [63, 64]). In addition, in somatic cells of mice, rats, and humans, a progressive accumulation of mtDNA aberrations (e.g., deletions, base substitutions) is observed during aging [65, 66] (for reviews see [63, 64]). The occurrence of both altered and unaltered mtDNA molecules and their subsequent segregation was suggested to result in tissue mosaics leading to tissues with various degrees of defective energetic capacities [67, 68]. In post-mitotic cells, the mutations accumulate, since no segregation occurs which divides the altered mtDNA molecules among the daughter cells. Therefore also in post-mitotic cells an increased rate of mtDNA mutation might lead to a subsequent functional impairment of the mitochondrial energy-producing apparatus.
GENETIC CONTROL OF GENOME INTEGRITY
The above mentioned examples indicate that considerable DNA rearrangements occur during aging of biological systems. As a whole, during lifespan, the genetic information is far from being stabile but undergoes a series of changes. Up to a certain extent, these changes may be tolerable, perhaps even advantageous if they would lead to a higher adaptability to altered environmental conditions. However, if the extent of these changes exceeds a critical threshold, the negative consequences may prevail, leading to degeneration and death.
Interestingly, in some cases, certain types of DNA reorganizations do not appear to occur accidentally but rather are resulting from programmed processes. Unfortunately, the details of such programmed changes are poorly understood although in some cases first ideas have emerged. In general, it is clear and almost trivial that genes exist which are involved in the control of the integrity of the genome. A number of such genes have been identified, though only in a few cases the activity of this type of genes has been correlated with aging (for review see [69]).
Telomeres
As mentioned above, in some systems telomere length is correlated with senescence. At least four different models have been put forward which correlate telomere length and aging processes (see Fig. 1).
One model describes senescence as a consequence of the deletion of essential genes located in the proximity of telomeres. During every replication cycle the chromosome ends shorten until a chromosome region becomes deleted in which essential genes are located. The loss of these genes leads to senescence [11] (for review see [70]). The second model [71] is principally similar, but more specific, suggesting the activity of a senescence repressor gene which is located in the proximity of the telomere. After substantial shortening of the telomeres this repressor becomes inactivated leading to senescence. Both models propose a direct influence of telomere length on the expression of adjacent genes. In principle, such a distance-dependent telomere position effect (TPE) has been described in S. cerevisiae [72]. However, in contrast to the inactivation of a senescence repressor, as postulated by model two, chromosome shortening leads to decreased silencing of functions and thereby rather to activation of adjacent genes. Unfortunately, investigations of a human cell clone which was transfected with a reporter gene that was targeted to a telomere region gave no evidence for such an effect [73]. Subclones of the original transgenic clone with different telomere lengths did not differ in the expression of the reporter gene.Fig. 1. Models explaining the influence of telomere length on aging processes. a) Model 1: senescence is described as a result of the deletion of essential genes located in the proximity of a telomere. b) Model 2: in juvenile cells a gene which codes for a senescence repressor and is located in the telomere region is expressed, leading to a suppression of a senescence pathway. Telomere shortening leads to the inactivation of this gene and the activation of the corresponding pathway. c) Model 3: a telomere-binding protein (TBP) is bound to the telomeres of germ cells and young somatic cells. The TBP gene is expressed at low levels. In senescent somatic cells TBP becomes released from telomeres and binds to the TBP gene region and a putative gene coding for a mitotic repressor. These two genes now are expressed at higher levels. d) Model 4: the expression of the TBP gene is influenced by telomerase. In germ line cells the expression is repressed to a low level. In somatic cells the TBP gene is not repressed due to the lack of telomerase. Free TBP leads to the activation of a mitotic repressor gene and as a consequence to senescence.
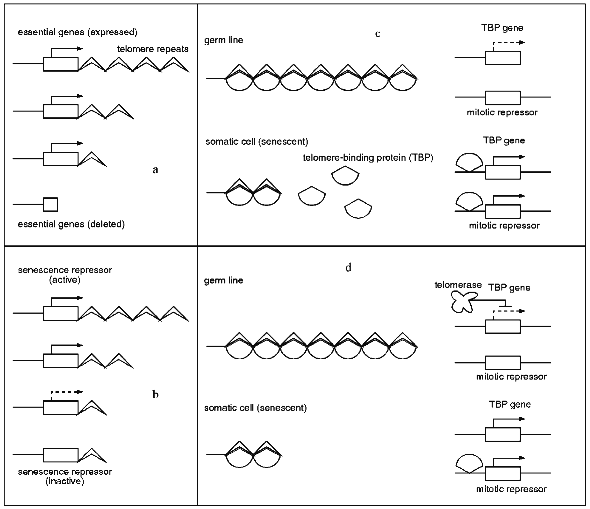
Several recent findings indicate a role of telomere-binding proteins in senescence. The putative role of these proteins is part of models 3 and 4, respectively (according to [16]). In model 3, the telomere-binding protein (TBP) binds to the telomere repeats. The amount of unbound TBP is a measure of telomere length. In the germ line and in somatic cells which went only through a few cell cycles virtually all TBPs are bound to the telomeres. In contrast, in aging cells with shortened telomeres increasing amounts of free, unbound TBP are found. The free protein may now be capable of raising its own synthesis via autoregulation. In addition, it is available to bind at other chromosomal regions leading to the expression of a mitotic repressor which contributes to senescence processes.
Alternatively, model 4 proposes a mechanism in which the amount of free TBP is not determined by the telomere length but rather by the activity of telomerase. In germ line cells, telomerase is suggested to inhibit the expression of the TBP gene. As a result, TBP is unable to activate the mitotic repressor. On the other hand, in aged somatic cells, telomerase activity is lost. Consequently, the concentration of TBP continuously increases until the mitotic repressor becomes activated, leading to senescence. In such a scenario, telomere shortening is a by-product resulting from a lack of telomerase activity in older somatic cells.
In support of this idea, a few telomere-binding proteins were characterized recently (for summary see [74]). Rap1p in S. cerevisiae binds to telomeric repeats and inhibits thereby the elongation of the telomeres. In addition to the binding sites in the telomeres, several binding sites are located upstream of genes for protein synthesis and glycolytic enzymes. The expression of these genes is mediated by protein kinase A [75] and may be involved in aging processes.
Quite recently, also in Schizosaccharomyces pombe (Taz1) and in humans (TRF1) two genes encoding telomere-binding proteins have been isolated [76, 77]. At least in S. cerevisiae, S. pombe, and in some immortal human cell lines these proteins limit the telomere elongation presumably by inhibition of telomerase. Interestingly, all three proteins share a myb-DNA-binding domain, although the rest of the protein sequence shows little homology. It may be speculated that these proteins are somehow involved in triggering aging processes via binding to the upstream region of different genes.
Nuclear Genes Affecting Lifespan
1. Saccharomyces cerevisiae. Due to the well developed methodologies to manipulate S. cerevisiae a number of age-relevant genes of this aging model have been isolated and characterized in the past several years (for review see [78, 79]). In one approach, genes were isolated which were expressed during different phases in lifespan. Among others, D´Mello et al. [80] isolated a gene termed LAG1 (longevity assurance gene). Deletion of the coding region of LAG1 led to a 50% increase in lifespan. The protein sequence of the putative LAG1 protein suggests that it possesses membrane-spanning domains. It shows no other significant homologies to other proteins.
In a second approach, the candidate gene approach, the two yeast RAS-encoding genes, RAS1 and RAS2,were demonstrated to influence yeast lifespan. While partial deletion of RAS1 leads to an increased lifespan, disruption of RAS2 results in a decrease [81]. Interestingly, overexpression of RAS2 leads to a 40% increase in lifespan and overexpression of RAS1 shows no effect.
Finally, genes affecting lifespan of yeast were isolated from mutants with an increased stress resistance. Among 39 mutants analyzed, eight also displayed an 20-55% increase in lifespan [82]. One of these mutants, uth 2-42 (uth for youth), was further investigated. It turned out that the mutant is characterized by a 50% increased lifespan. In addition, it mates poorly and exhibits a polar budding pattern which normally is characteristic not for haploid but for diploid yeast. The gene was cloned and found to code for SIR4 (silent information regulator), a well known gene coding for one component of the yeast silencing complex which is involved in the silencing at the silent mating type loci (HML and HMR). The mutation of the uth 2-42 mutant (later called SIR4-42) results in a truncated protein, which lacks 121 amino acids at the C-terminus. On the basis of these data, the authors suggested a model in which the mutated SIR4 protein, together with SIR2 and SIR3, is not able to silence the Rap1p-binding loci (the telomeres and HM-loci). Instead, the altered SIR-complex was proposed to silence the so-called age locus, a new gene suggested to be involved in determining yeast lifespan [82]. In a subsequent paper [83], the age locus was identified as the rDNA locus, a locus that has long been discussed, although controversially, to be involved in aging in various biological systems (see above). It was found that during aging of yeast cells the SIR complex becomes relocated from the telomeres to the nucleolus, where the rDNA locus is located. However, the exact function of the SIR complex during aging of the wild type and the SIR4-42 remains to be elucidated. Silencing of age (rDNA locus), as originally thought, does not appear to play a role [83].
2. Filamentous Fungi. Also in filamentous fungi, a number of genes are known to affect lifespan. This became first evident from a number of long-lived mutants. Because of technical problems the cloning of these genes is not so easy as in S. cerevisiae. However, in the meantime, a few relevant genes have been cloned and analyzed. Interestingly, some of these genes appear to code for components of a molecular apparatus that links nuclear encoded genes and mitochondrial functions. In particular, age-related mtDNA instabilities seem to be controlled with some of these genes.
In the filamentous fungus Neurospora crassa, Seidel-Rogol et al. [84] reported that a nuclear mutant, nd (natural death), which displays a premature death syndrome is characterized by unstable mtDNA. The mutated gene seems to be of crucial importance for the maintenance of the mtDNA. The wild-type nd gene was suggested to contribute to replication, repair or recombination processes.
In Podospora anserina, a number of nuclear mutants with an altered lifespan have been selected and initially analyzed (see Table 1). Among these, mutations affecting translation fidelity were found to have an influence on lifespan. Recently, one of these genes, AS4, was identified as translation elongation factor EF-1alpha [85]. Another nuclear gene involved in the control of aging in P. anserina, gerontogene grisea, was cloned utilizing a complementation approach [86]. The corresponding mutant is a pleiotropic mutant with an altered morphology (e.g., change in ascospore color) and an increase in lifespan of about 56% [87, 88]. Interestingly, the combination of the mutant copy of grisea with another mutant gene, vivax (viv), has a synergistic effect leading to an immortal double mutant (for review see [41]). Among others, these data indicate that different nuclear genes are involved in the control of aging in P. anserina. The cloning and characterization of the wild type and the mutant copy of grisea revealed that it codes for a transcription activator with significant homology to the ACE1/MAC1/AMT1 family of Saccharomyces cerevisiae and Candida glabrata, respectively [89-91] (for review see [92]). One out of four point mutations identified in the cloned gene copy of the long-lived mutant leads to a splicing defect. The long-lived mutant thus is a loss-of-function mutant in which no functional GRISEA transcription factor is produced. Interestingly, further investigations revealed that the phenotype of mutant grisea, including lifespan, can be rescued by adding higher amounts of copper to the growth medium than usual [93]. This finding and the fact that the three homologs of S. cerevisiae and C. glabrata are copper-dependent transcription activators involved in a tight control of cellular copper homeostasis also points to an effect of cellular copper on lifespan. Since it is known that copper can lead to the generation of the hydroxyl free radical via Fenton chemistry [94] the analyses of long-lived mutant grisea links aging of this simple system also to reactive oxygen species and the free radical theory of aging [95, 96]. In addition, in P. anserina now a view is emerging indicating that aging is controlled by a complex network of mitochondrial--nuclear interactions. One component of this network is transcription activator GRISEA which appears to be involved in the control of the age-specific mitochondrial DNA rearrangement discussed above [97].
TABLE 1. Nuclear Genes Associated with
Lifespan Prolongation in Podospora anserina (only those genes
are listed which have been initially characterized on the molecular
level)

3. Caenorhabditis elegans. In the nematode Caenorhabditis elegans several mutations with lifespan prolonging influence were investigated in the past several years (for review see [98, 99]). A recessive mutation of the age-1 gene was found to result in a 60% increased maximum lifespan at 20°C [100]. Dorman et al. [101] reported that the recessive mutations of age-1 and daf-2 have only a lifespan prolonging effect if daf-16 and daf-18 are active, suggesting a common genetic pathway. Recently, Murakami and Johnson [102] showed a correlation between UV resistance and longevity of a number of so-called Age mutants, age-1, daf-2, daf-23, spe-26, and clk-1. Mutation of the single daf-16 gene suppresses both UV resistance and longevity of all of these mutants suggesting a genetic pathway in which UV resistance and lifespan is negatively regulated be a group of gerontogenes. It appears likely that in Caenorhabditis elegans only very few if not only one genetic pathway leads to longevity and stress resistance. Here it will be of great interest to identify and further characterize the genes contributing to this pathway.
4. Humans. In humans, some data indicate the relevance of nuclear genes in the control of age-related mtDNA reorganizations. In one experimental study, Hayashi et al. [103] transferred mtDNA from donors of different age to mtDNA deficient HeLa cells. The mitochondria of the donors showed no decrease in the copy number of intact mtDNA molecules nor an increase in the copy number of defective mtDNA molecules. However, mitochondria of older donors were compromised showing a significant decrease in polypeptide synthesis and in cytochrome oxidase activity. Interestingly, the transfer experiments revealed that mtDNA from older donors is intact in the nuclear background of HeLa cells. Vice versa, transfer of HeLa nuclei into fibroblasts from aged donors restored mitochondrial protein synthesis and cytochrome oxidase activity. These results indicate that nuclear recessive mutations are related to the age-related mitochondrial dysfunction found in aging human skin fibroblasts. The identification of the affected nuclear genes will be of great benefit for the analysis of the molecular consequences of the mutations.
Recently, the cloning and initial characterization of a nuclear gene with considerable influence on lifespan was reported. Yu et al. [104] isolated the WRN gene, the mutation of which leads to Werner's syndrome, via positional cloning. As mentioned above, this recessive disease is characterized by extensive deletions and other DNA reorganizations [8]. The WRN gene was found to code for a putative helicase. Helicases contribute to a number of processes in DNA metabolism (e.g., replication, recombination, transcription, and repair) by their ability to unwind DNA. It is thought that an altered DNA metabolism is responsible for the symptoms and characteristics of Werner's syndrome. However, it remains unclear if the mechanisms underlying the expression of these characteristics correspond with those of normal aging [105].
Taking together, it is clear that one major general component involved in aging is related to age-dependent DNA reorganizations. Reorganizations in both genetic compartments of a eukaryotic cell, the nucleus and in mitochondria, are of significance. Apart from the accumulation of random genetic rearrangements, genetically programmed processes appear to be involved at least in some systems. In these cases, a rather complex molecular machinery encoded by different genes appears to be involved [106]. Currently, only a few of these genes have been cloned from different organisms and are under investigation. It is the challenge of the next years to identify, clone, and analyze further relevant genes of this type. Finally, the strategy to unravel the genetic basis of different biological systems, lower and higher organisms, appears to be promising because it can be expected to give rise to a detailed understanding of the basic biological process of aging and will answer a currently unsolved question to whether or not a unified mechanism of aging exists in all organisms.
The experimental work was performed within the framework of the Concerted Action Program (No. BMH1-CT94-1710) of the European Community and was supported by grants from the Deutsche Forschungsgemeinschaft (Bonn-Bad Godesberg, Germany).
LITERATURE CITED
1.Schneider, E. L. (1985) Handbook of the Biology
of Aging (Schneider, E. L., and Rower, J. W., eds.) Academic Press,
New York, pp. 357-373.
2.Abruzzo, M. A., Mayer, M., and Jacobs, P. A. (1985)
Cytogenet. Cell Genet., 39, 275-278.
3.Richard, F., Aurias, A., Couturier, J., Dutrillaux,
A. M., Flüry-Hérard, A., Gerbault-Seureau, M., Hoffschir, F.,
Lamoliatte, E., Lefrancois, D., Lombard, M., Muleris, M., Prieur, M.,
Ricoul, M., Sabatier, L., Viegas-Péquignot, E., Volobouev, V., and
Duttrillaux, B. (1993) Mutat. Res., 295, 71-80.
4.Griffin, D. K., Abruzzo, M. A., Millie, E. A.,
Sheean, L. A., Feingold, E., Sherman, S. L., and Hassold, T. J. (1995)
Human Mol. Genet., 4, 2227-2232.
5.Martin, G. M., Smith, A. C., Ketterer, D. J.,
Ogburn, C. E., and Disteche, C. M. (1985) Isr. J. Med. Sci.,
21, 296-301.
6.Marlhens, F., Al Achkar, W., Aurias, A., Couturier,
J., Dutrillaux, A. M., Gerbault-Seureau, M., Hoffschir, F., Lamoliatte,
E., Lefrancois, D., Lombard, M., Mulris, M., Prieur, M.,
Prod´homme, M., Sabatier, L., Viegas-Pequignot, E., Volobouev,
V., and Dutrillaux, B. (1986) Hum. Genet., 73,
290-297.
7.Bender, M. A., Preston, R. J., Leonard, R. C.,
Pyatt, B. E., and Gooch, P. C. (1989) Mutat. Res., 212,
149-154.
8.Fukuchi, K.-I., Martin, G. M., and Monnat, R. T.
(1989) Proc. Natl. Acad. Sci. USA, 86, 5893-5897.
9.Strehler, B. L. (1986) Exp. Gerontol.,
21, 283-319.
10.Gaubatz, J. W. (1990) Mutat. Res.,
237, 271-292.
11.Olovnikov, A. M. (1973) J. Theor. Biol.,
41, 181-190.
12.Harley, C. A., Futcher, A. B., and Greider, C. W.
(1990) Nature, 345, 458-460.
13.Counter, C. M., Avilion, A. A., LeFeuvre, C. E.,
Stewart, N. G., Greider, C. A., Harley, C. B., and Bacchetti, S. (1992)
EMBO J., 11, 1921-1929.
14.Harley, C. A., Vaziri, H., Counter, C. M., and
Allsopp, R. C. (1992) Exp. Gerontol., 27, 375-382.
15.Broccoli, D., Godley, L. A., Donehover, L. A.,
Varmus, H. E., and de Lange, T. (1996) Mol. Cell Biol.,
16, 3765-3772.
16.Kilian, A., Stiff, C., and Kleinhofs, A. (1995)
Proc. Natl. Acad. Sci. USA, 92, 9555-9559.
17.Walmsley, R. M., and Petes, T. D. (1985) Proc.
Natl. Acad. Sci. USA, 82, 506-510.
18.Jazwinski, S. M. (1996) Handbook of the
Biology of Aging (Schneider, E. L., and Rowe, J. W., eds.) Academic
Press, New York, pp. 39-54.
19.Li, B., and Lustig, A. J. (1996) Genes
Dev., 10, 1310-1326.
20.Schwartz, T., and Osiewacz, H. D. (1996)
Mutat. Res., 316, 193-199.
21.Murray, V. (1990) Mutat. Res.,
237, 59-63.
22.Nikitin, A. G., and Shmookler Reis, R. J. (1997)
Genet. Res. Camb., in press.
23.Osiewacz, H. D., and Heinen, U. (1989) Progr.
Botany, 50, 174-197.
24.McClanahan, T. A., and McEntee, K. (1984) Mol.
Cell. Biol., 4, 2356-2363.
25.Bradshaw, V. A., and McEntee, K. (1989) Mol.
Gen. Genet., 218, 465-474.
26.Egilmez, N. K., and Shmookler Reis, R. J. (1994)
Mutation Res., 316, 17-24.
27.Levis, R. W., Ganesan, R., Houtchens, K., Tolar,
L. A., and Sheen, F. (1993) Cell, 75, 1083-1093.
28.Biessmann, H., Mason, J. M., Ferry, K.,
d´Hulst, M., Valgeirsdottir, K., Traverse, K. L., and Pardue,
M.-L. (1990) Cell, 61, 663-673.
29.Biessmann, H., Valgeirsdottir, K., Lofsky, A.,
Chin, C., Ginther, B., Levis, R. W., and Pardue, M.-L. (1992) Mol.
Cell. Biol., 12, 3910-3018.
30.Woodruff, R. C. (1992) Genetica,
86, 143-154.
31.Woodruff, R. C., and Nikitin, A. G. (1995)
Mutat. Res., 338, 35-42.
32.Schmid, C. W., and Jelinek, W. R. (1982)
Science, 216, 1065-1070.
33.Servomaa, K., and Rytömaa, T. (1988) Cell
Tissue Kinet., 21, 33-43.
34.Servomaa, K., and Rytömaa, T. (1990) Int.
J. Radiat. Biol., 57, 331-345.
35.Esser, K. (1984) in The Sandoz Lectures in
Gerontology (Bergener, M., Ermini, M., and Stähelin, H. B.,
eds.) Academic Press, London, pp. 3-20.
36.Kück, U. (1989) Exp. Mycol.,
13, 111-120.
37.Osiewacz, H. D. (1995) in Molecular Aspects of
Aging (Esser, K., and Martin, G. M., eds.) Wiley, Chichester, UK,
pp. 29-44.
38.Osiewacz, H. D. (1996) in Fungal Genetics
(Bos, C. J., ed.) Dekker, New York, pp. 317-335.
39.Rizet, G. (1953) C. R. Acad. Sci. Paris,
237, 838-840.
40.Marcou, D. (1961) Ann. Sci. Natur Bot.
Ser., 12, 653-764.
41.Esser, K., and Tudzynski, P. (1980) in
Senescence in Plants (Thimann, K. V., ed.) CRC Press, Boca
Raton, pp. 67-83.
42.Stahl, U., Lemke, A., Tudzynski, P., Kück,
U., and Esser, K. (1978) Mol. Gen. Genet., 162,
341-343.
43.Cummings, D. J., Belcour, L., and Grandchamp, C.
(1979) Mol. Gen. Genet., 172, 239-250.
44.Osiewacz, H. D., and Esser, K. (1984) Curr.
Genet., 8, 299-305.
45.Cummings, D. J., McNally, I. A., Domenico, J. M.,
and Matsuura, E. T. (1985) J. Mol. Biol., 185,
659-680.
46.Belcour, L., Begel, O., Mossé, M. O., and
Vierny, C. (1981) Curr. Genet., 3, 13-21.
47.Kück, U., Stahl, U., and Esser, K. (1981)
Curr. Genet., 3, 151-156.
48.Sellem, C. H., Lecellier, G., and Belcour, L.
(1993) Nature, 366, 176-178.
49.Faßbender, S., Brühl, K. H., Ciriacy,
M., and Kück, U. (1994) EMBO J., 13, 2075-2083.
50.Jamet-Vierny, C., Begel, O., and Belcour, L.
(1980) Cell, 21, 189-194.
51.Jamet-Vierny, C., Boulay, J., Begel, O., and
Silar, P. (1997) Curr. Genet., 31, 171-178.
52.Griffiths, A. J. F. (1992) Annu. Rev.
Genet., 26, 351-357.
53.Akins, R. A., Kelley, R. L., and Lambowitz, A. M.
(1986) Cell, 47, 505-516.
54.Bertrand, H., Chan, B. S.-S., and Griffiths, A.
J. F. (1985) Cell, 41, 877-884.
55.Chan, B. S.-S., Court, D. A., Vierula, P. J., and
Bertrand, H. (1991) Curr. Genet., 20, 225-237.
56.Court, D. A., Griffiths, A. J. F., Kraus, S. R.,
Russell, P. J., and Bertrand, H. (1991) Curr. Genet.,
19, 129-137.
57.Osiewacz, H. D., Hermanns, J., Marcou, D.,
Triffi, M., and Esser, K. (1989) Mutat. Res., 219,
1-7.
58.Hermanns, J., and Osiewacz, H. D. (1992) Curr.
Genet., 22, 491-500.
59.Hermanns, J., Asseburg, A., and Osiewacz, H. D.
(1994) Mol. Gen. Genet., 243, 297-307.
60.Hermanns, J., and Osiewacz, H. D. (1996) Curr.
Genet., 29, 250-256.
61.Koll, F., Boulay, J., Belcour, L., and
d´Aubenton-Carafa, Y. (1996) Nucleic Acids Res.,
24, 1734-1741.
62.Jamet-Vierny, C., Boulay, J., and Briand, J.-F.
(1997) Curr. Genet., 31, 162-170.
63.Osiewacz, H. D., and Hermanns, J. (1992)
Aging, 4, 273-286.
64.Wallace, D. C. (1995) in Molecular Aspects of
Aging (Esser, K., and Martin, G. M., eds.) Wiley, Chichester, UK,
pp. 163-177.
65.Piko, L., Hougham, A. J., and Bulpitt, K. J.
(1988) Mech. Ageing Dev., 43, 279-293.
66.Hayakawa, M., Katsumata, K., Yoneda, M., Tanaka,
M., Sugiyama, S., and Ozawa, T. (1996) Biochem. Biophys. Res.
Commun., 226, 369-377.
67.Linnane, A. W., Marzuki, S., Ozawa, T., and
Tanaka, M. (1989) Lancet, 1, 642-645.
68.Kadenbach, B., and Höcker-Müller, J.
(1990) Naturwissenschaften, 77, 221-225.
69.Osiewacz, H. D. (1997) J. Mol. Med., in
press.
70.Harley, C. A. (1991) Mutat. Res.,
256, 271-282.
71.Wright, W. E., and Shay, J. W. (1992) Trends
Genet., 8, 193-197.
72.Gottschling, D. E., Aparicio, O. M., Billington,
B. L., and Zakian, V. A. (1990) Cell, 63, 751-762.
73.Sprung, C. N., Sabatier, L., and Murnane, J. P.
(1996) Nucleic Acids Res., 24, 4336-4340.
74.Shore, D. (1997) Nature, 385,
676-677.
75.Klein, C., and Struhl, K. (1994) Mol. Cell.
Biol., 14, 1920-1928.
77.Cooper, J. P., Nimmo, E. R., Allshire, R. C., and
Cech, T. R. (1997) Nature, 385, 744-747.
76.Van Steensel, B., and de Lange, T. (1997)
Nature, 385, 740-743.
78.Jazwinski, S. M. (1990) J. Gerontol.,
45, B68-B74.
79.Jazwinski, S. M. (1995) in Molecular Aspects
of Aging (Esser, K., and Martin, G. M., eds.) Wiley, Chichester,
UK, pp. 15-28.
80.D´Mello, N. P., Childress, A. M., Franklin,
D. S., Kale, S. P., Pinswasdi, C., and Jazwinski, S. M. (1994) J.
Biol. Chem., 22, 15451-15459.
81.Sun, J., Kale, S. P., Childress, A. M.,
Pinswasdi, C., and Jazwinski, S. M. (1994) J. Biol. Chem.,
28, 18638-18645.
82.Kennedy, B. K., Austriaco, N. R., Jr., Zhang, J.,
and Guarente, L. (1995) Cell, 80, 485-496.
83.Kennedy, B. K., Gotta, M., Sinclair, D. A.,
Mills, K., McNabb, D. S., Murthy, M., Pak, S. M., Laroche, T., Gasser,
S. M., and Guarente, L. (1997) Cell, 89, 381-391.
84.Seidel-Rogol, B. L., King, J., and Bertrand, H.
(1989) Mol. Cell Biol., 9, 4259-4264.
85.Silar, P., and Picard, M. (1994) J. Mol.
Biol., 235, 231-236.
86.Osiewacz, H. D., and Nuber, U. (1996) Mol.
Gen. Genet., 252, 115-124.
87.Prillinger, H., and Esser, K. (1977) Mol. Gen.
Genet., 156, 333-345.
88.Tudzynski, P., and Esser, K. (1979) Mol. Gen.
Genet., 173, 71-84.
89.Thiele, D. (1988) Mol. Cell. Biol.,
6, 1158-1163.
90.Zhou, P., and Thiele, D. J. (1991) Proc. Natl.
Acad. Sci. USA, 88, 6112-6116.
91.Jungmann, J., Reins, H. A., Lee, J., Romeo, A.,
Hassett, R., Kosman, D., and Jentsch, S. (1993) EMBO J.,
12, 5051-5056.
92.Zhou, P., and Thiele, D. J. (1993)
BioFactors, 4, 105-115.
93.Marbach, K., Fernández-Larrea, J., and
Stahl, U. (1994) Curr. Genet., 26, 184-186.
94.Halliwell, B., and Gutteridge, M. C. (1984)
Biochem. J., 219, 1-14.
95.Harman, D. (1956) J. Gerontol.,
11, 298-300.
96.Harman, D. (1983) Age, 6,
86-94.
97.Borghouts, C., Kimpel, E., and Osiewacz, H. D.,
submitted.
98.Lithgow, G. (1996) in Handbook of the Biology
of Aging (Schneider, E. L., and Rowe, J. W., eds.) Academic Press,
New York, pp. 39-73.
99.Smeal, T., and Guarente, L. (1997) Curr. Opin.
Genet. Dev., 7, 281-287.
100.Friedman, D. B., and Johnson, T. E. (1988)
Genetics, 118, 75-86.
101.Dorman, J. B., Albinder, B., Shroyer, T., and
Kenyon, C. (1995) Genetics, 141, 1399-1406.
102.Murakami, S., and Johnson, T. E. (1996)
Genetics, 143, 1207-1218.
103.Hayashi, J.-I., Ohta, S., Kagawa, Y., Kondo,
H., Kaneda, H., Yonekawa, H., Takai, D., and Miyabayashi, S. (1994)
J. Biol. Chem., 269, 6878-6883.
104.Yu, C. E., Oshima, J., Fu, Y. H., Wijsman, E.
M., Hisama, F., Alisch, R., Matthews, S., Nakura, J., Miki, T., Ouais,
S., Martin, G. M., Mulligan, J., and Schellenberg, G. D. (1996)
Science, 272, 258-262.
105.Epstein, C. J., and Motulsky, A. G. (1996)
BioEssays, 18, 1025-1027.
106.Tudzynski, P., Stahl, U., and Esser, K. (1982)
Curr. Genet., 6, 219-222.