Critical Telomere Shortening Regulated by the Ataxia--Telangiectasia Gene Acts as a DNA Damage Signal Leading to Activation of p53 Protein and Limited Life-Span of Human Diploid Fibroblasts. A Review
H. Vaziri1
1Ontario Cancer Institute, 610 University Avenue, 9-321 Toronto, Ontario, M5G-2M9 Canada; fax: 416-946-2065; E-mail: Vaziri@oci.utoronto.ca
Submitted August 14, 1997.
Somatic cells undergo a limited number of doublings in culture and enter an irreversible block in the G1 and G2/M phase of the cell cycle termed "senescence". Telomere shortening presumably as a consequence of the end-replication problem has been proposed to act as a mitotic clock eventually leading to cellular senescence. Several models have been proposed to explain how telomere shortening can lead to cellular senescence. We proposed previously that telomere shortening may eventually lead to formation of dicentric chromosomes which on subsequent breakage activate a DNA damage response pathway involving the p53 protein. Hence we proposed that the telomere shortening signal is perceived by the cell as DNA damage. Recently we have obtained experimental evidence that the p53 protein is activated posttranslationally in human fibroblasts which undergo telomere shortening and subsequent senescence in culture. In this paper we also show that the increased activity of p53 protein coincides with formation of dicentric chromosomes and senescence. Also, we have previously found that an increase in the level of the down stream target of p53 protein, p21WAF1/SDI1/CIP1, is dependent on both p53 and p300 proteins. We have also shown that fibroblasts obtained from individuals with Ataxia Telangiectasia lose telomeric DNA at an accelerated rate, activate p53 protein, and undergo premature senescence in culture. These results suggest that the ataxia--telangiectasia gene (ATM) and p53 are involved in surveillance and regulation of telomeric DNA. Once a critical length of telomeric DNA is reached, ATM and p53 sense and relay this signal to the cell cycle leading to senescence.
KEY WORDS: p53, p21, ATM, senescence, telomere, aging, fibroblasts, DNA damage.
Senescence, Telomeres, and the End-Replication Problem
Normal human diploid fibroblasts have a limited life-span and undergo senescence [1]. Senescent cells are viable and are arrested in G1/S and G2/M phases of the cell cycle [2]. Vertebrate chromosomes terminate in tandem repeats of (TTAGGG)n which form highly conserved nucleoprotein structures called "telomeres". These structures are believed to protect the ends of chromosomes against exonucleases and illegitimate recombination. Their most important function, however, may be to act as a buffer zone against the "end-replication problem". All DNA polymerases require a free 3´-OH and a DNA template in order to synthesize DNA from the 5´ to 3´ direction. The 3´-OH is provided on the lagging strand by Okazaki fragments which act as primers for DNA synthesis. After each round of replication, terminal fragments of DNA are left unreplicated due to the removal of the terminal Okazaki fragments and the inability to prime DNA synthesis from the free DNA ends. The end-replication problem was first proposed by Aleksey Olovnikov in 1971 [3], as a mechanism which restricts the life-span of human cells.
Telomere Hypothesis of Cell Aging and Immortalization
Two decades after the proposal of the marginotomy theory by Olovnikov, Harley and coworkers discovered that eventual loss of telomeric DNA as a function of cell division may be involved in cellular senescence [4-6]. Most senescent cells have approximately the same amount of telomeric DNA, suggesting that a critical length of telomeric DNA may eventually signal the G1 arrest at senescence [7]. Moreover, the telomere length is correlated significantly with the replicative potential of human fibroblasts [8]. Loss of telomeric DNA is observed in variety of hematopoietic cell types in vivo which may contribute to immunosenescence [9, 10].
The telomere hypothesis proposes that telomere loss acts as a mitotic clock which counts the number of cell divisions. A critical length of telomeric DNA will then trigger the cell cycle arrest. When cells are transfected with SV40 LT antigen they will bypass senescence and continue to lose telomeric DNA until crisis. Overcoming crisis is associated with stabilized telomeres and expression of telomerase [11], a ribonucleoprotein reverse transcriptase capable of elongating telomeres in vivo [12]. It has been shown that telomerase is activated in most cell lines and primary tumors tested, but not in normal cells [13]. Hence telomerase expression is considered to be essential for cell immortality. The recent discovery of post-crisis cells and tumor lines lacking telomerase expression which maintain telomere length through an alternative pathway (ALT) [14, 22], however, suggests that telomerase activity may be dispensable for cell survival.
Telomere Loss or DNA Damage?
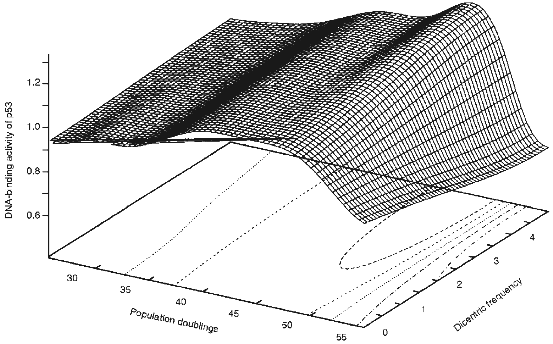
The mechanism by which telomere shortening leads to cell cycle arrest in G1 is unknown. Based on the observation that dicentric and ring chromosomes are seen at a relatively high frequency (30-70%) at senescence and a short critical telomere length is reached, we wished to propose a third model, "the telomere loss/DNA-damage hypothesis" [15], in which the loss of telomeric DNA from any subset of chromosomes generates dicentric and ring chromosomes and eventual breakage during the next mitosis generates a signal which activates a "DNA damage" response pathway culminating in G1 arrest and senescence. As shown in Fig. 1, the frequency of dicentric chromosomes is dramatically increased near senescence and coincides with the activation of p53 protein. This suggests that formation of dicentric chromosomes and their subsequent breakage activates p53 protein.
p53 Protein and DNA DamageFig. 1. 3D representation of the senescence-associated DNA damage (SAD) peak in HDFs (human diploid fibroblasts). DNA-binding activity of p53 protein was measured during passaging of the cells and data was plotted against the dicentric frequency [30]. The data were simulated and graphed using GNUPLOT.
p53 protein is a transcription factor known to mediate a G1 arrest in response to DNA damaging agents. However, both p53-dependent and p53-independent pathways may exist to control the G1--S transition in human diploid fibroblasts (HDFs). Multiple targets have been identified down stream of p53, some of which include p21 [16], Gadd45, Mdm2, and cyclin G. p21 has also been shown to be a potent inhibitor of CDK2,4-cyclin-A,E-kinase activity [17]. Recently it was also shown that gamma irradiation of HDFs leads to induction of p53 and p21 and a G1 block. Interestingly, these cells also show morphological changes reminiscent of cellular senescence [18]. Most interestingly, p21 was also shown to be upregulated in senescent human fibroblasts [19].
p53 Protein and Cell Aging
It has been suggested that p53 is a senescent associated gene [20]. However their conclusions were based on experiments in which cells, transfected with LT antigen or E6/E7, escaped senescence and had an extension of life-span. In addition, extension of the life-span of HDFs from individuals with Li--Fraumeni syndrome which have a germline p53 mutation has been observed [21]. Furthermore, this extension of life-span is associated with loss of p53 [22]. In light of the role of p53 in regulating the G1 checkpoint [23] and the fact that in all of the above experiments, p53 was inactivated in young passage cells, escape from senescence could have been due to genomic instability and secondary mutations associated with loss of wild-type p53 function. The fact that overexpression of mutant p53 in normal cells can also extend cellular life-span [24, 25] suggests a more direct role for p53 in cellular senescence. Earlier studies showed that there is no increase in the level of mRNA of p53 during senescence [26]. Other studies have shown either no change [27] or an increase in the level of p53 protein in senescent cells [28]. Our studies indicate that the discrepancies observed between these results are due to the methods used to detect p53 protein. We have shown that there is no change in the steady-state level of p53 protein when measured by direct western blot analysis [29]. However, when p53 protein is immunoprecipitated and then subsequently detected by a secondary antibody, different isoforms of the protein are detected [29], which makes it appear as an overexpression. Different isoforms of p53 protein seem to accumulate in aging cells despite the lack of an increase in the net steady state of the protein. Such alternative forms of p53 protein may activate different down stream growth inhibitory pathways including p21 and IGF-BP3, which would in turn contribute to initiation of cellular senescence.
Increased DNA-Binding Activity of p53 Protein during Senescence
Despite no net change in the level of p53 protein during cell aging, there is a moderate increase in the DNA-binding activity of p53 protein. Using a labelled p53 consensus sequence we have shown that there is an increase in the DNA-binding activity of p53 protein independent of the level of the protein [29]. Interestingly, it has been shown previously that near senescent cells accumulate a significant number of dicentric and ring chromosomes [30]. It is possible that the critical telomere shortening in such cells lead to accumulation of such chromosomes. Subsequent breakage of the dicentric chromosomes in the next mitosis will lead to generation of at least one double-strand or single-strand break. Such breaks are then perceived by the cells as a DNA damage signal which would lead to activation of p53 protein and subsequent G1 arrest [29]. When the increase in the DNA-binding activity of p53 protein is plotted versus the age and number of dicentrics, the presence of a SAD (Senescence-Associated DNA Damage) peak becomes evident [29]. When such data is simulated as a 3D representation (Fig. 1), the presence of the SAD peaks becomes even more clear.
Overexpression of p21 at Senescence is p53- and p300-Dependent
Both p53 dependent and independent pathways are known to activate p21 in response to differentiation or DNA damage [31]. We asked the question whether the induction of p21 observed at cellular senescence is dependent on p53 or other factors. We demonstrated that this induction is partially dependent on both p53 and p300 [29]. p300 is known to be both a transcriptional co-activator and a histone acetyltransferase [32]. Hence, it is possible that p300 is involved in the facilitation of transcription by opening the histone core and hence rendering the DNA more accessible to a RNA polymerase complex and transcriptional enhancers like p53.
ATM Regulates Telomere Length and Cellular Life-Span
Ataxia--telangiectasia (AT) is an autosomal recessive genetic disease characterized by the progressive cerebellar ataxia resulting in loss of the Purkinje cells of the cerebellum, dilated blood vessels (telangiectases), immune deficiency, increased chromosomal aberrations, cancer proneness, and sensitivity to ionizing radiation and radiomimetic drugs [33]. The AT gene designated ATM is mapped to 11q22-23 [34]; it has recently been cloned [35]. The 13 kb transcript containing 65 exons produces a predicted 350 kD protein and shows homology to a family of phosphatidylinositol-3-kinases (PI-3-kinase). Among these proteins which are involved in DNA damage signalling and cell cycle control are TEL1p [36], MEC1p [37] in S. cerevisiae, TOR proteins (yeast and mammalian), FRAP, mei-41 in D. melanogaster [38], and the catalytic subunit of DNA-dependent protein kinase (DNA-PK) in mammals [39]. Mutation in these genes leads to a variety of phenotypes similar to that of AT, including sensitivity to ionizing radiation, chromosomal instability, and cell cycle check point defects. Most interestingly inactivation of TEL1p, the yeast homology of AT, leads to progressive telomere shortening in yeast [36]. It has been shown previously that fibroblasts derived from patients with AT have a reduced life-span and undergo premature senescence [40]. Also it has been shown recently that the lengths of telomeric restriction fragments are shorter in AT lymphocytes [41] and human fibroblasts undergoing senescence [29]. Moreover, we showed that lack of ATM leads to an accelerated loss of telomeric DNA as a function of population doublings [29]. These data are consistent with the idea that AT is involved in the regulation of telomere length in humans. It has been proposed that the p53-dependent G1 check point in response to radiation is defective in AT cells [42]. Based on this preliminary evidence obtained from EBV-transformed B-cells, it has been proposed that AT acts upstream of p53 and is involved in its regulation/activation in response to DNA damage. In contrast to these results, the level and transactional activity of p53 protein in response to certain radiomimetic drugs like cisplatin are intact in AT cells when compared to normal controls [43]. Further investigation has also shown that the p53-dependent checkpoint in response to ionizing radiation is intact and only somewhat delayed in fibroblast strains derived from AT patients [44].
We provided further evidence that the increase observed in the DNA-binding activity of p53 protein during senescence is preserved in fibroblasts derived from AT patients [29]. These results suggest that lack of ATM may have in fact two separate effects on HDFs: 1) lack of ATM leads to an accelerated loss of telomeric DNA which eventually leads to senescence; 2) since ATM and p53 are known to interact [45], binding of ATM to p53 could potentially lead to inactivation of p53. In AT fibroblasts which lack the ATM protein this interaction is abrogated, leading to the hyperactivation of p53 protein in senescent cells. It should be noted that these scenarios may not be mutually exclusive considering the severe premature senescence of AT cells.
Emergence of a Coherent Model for Cellular Aging
Recent advances in understanding tumor suppressor genes and cell cycle control and telomere biology has led to formation of the "telomere loss/DNA damage hypothesis" for cell aging [15]. Long telomeres which are regulated by a complex of several proteins, do not normally lead to a cell cycle arrest. Loss of a critical amount of telomeric DNA can occur due to inactivation of a protein involved in regulation (like ATM) of telomeres. Critical shortening of telomeres can also occur due to the end-replication problem or other factors. Critically short telomeres in near senescent cultures of HDFs lead to formation of dicentric chromosomes. These chromosomes are broken in the next round of mitosis and this breakage would lead to activation of p53 protein and subsequent overexpression of p21 and IGF-BP3. p21 could bind to CDK2 and CDK4 and inhibit their activity. This could lead to failure of pRB to get phosphorylated which is required for progression through G1. On the other hand, proteins like IGF-BP3 could work in an autocrine-paracrine loop by binding to IGFs and hence neutralize the mitogenic activity of growth factors. This could partially explain the loss of response of senescent cells to serum and growth factors. Overexpression of other proteins such as p16 could also lead to hypophosphorylation of RB and failure to progress through G1. Together all such redundant mechanisms are required to ensure senescence and hence limited life-span of human cells. The senescence-associated DNA damage (SAD) (Fig. 1) may provide a plausible threshold mechanism which once triggered leads to the activation of the cellular senescence program. We propose that the p53 protein is the upstream threshold sensor which once activated initiates the senescence cascade in response to critical telomere shortening.
Relevance to Neoplastic Transformation
Loss of p53 function in a large majority of human cancers has put forward the paradigm that loss of p53 function in normal cells leads to a destabilized genome and a mutator phenotype which eventually leads to accumulation of further genetic change and subsequent immortalization of human cells. In the light of the new role of p53 in regulating cellular aging and life-span, it becomes clear that loss of p53 function due to mutation or deletion would lead to bypass of senescence. This would lead to an extension of the life-span until a second phase of arrest named "crisis" is reached. Once cells have bypassed crisis through additional genetic change and activation of telomere maintenance mechanisms, they acquire immortality and presumably a mutator phenotype. Hence, the early events which lead to inactivation of p53 protein may lead to formation of tumors in the absence of minimal genetic change merely through the extension of life-span of the normal cells. This is in opposition to the current paradigms that formation of tumors are due to a sequence of activation-inactivation of a multiple set of genes.
There is no doubt that once the cells have bypassed crisis and acquired immortality, they will go through even more genetic changes including those required for metastasis. The most important early events which lead to formation of tumors in vivo may not depend on multiple genetic changes, but only few changes which lead to extension of life-span of normal cells through inactivation of senescence genes. This would predict a better prognosis for tumors which are in their extended life-span (pre-crisis) phase than those which are post-crisis and immortal.
LITERATURE CITED
1.Hayflick, L., and Moorhead, P. (1961) Exp. Cell
Res., 25, 585-621.
2.Sherwood, S. W., Rush, D., Ellsworth, J. L., and
Schimke, R. T. (1988) Proc. Natl. Acad. Sci. USA, 85,
9086-9090.
3.Olovnikov, A. M. (1971) Dokl. Akad. Nauk
SSSR, 201, 394-397.
4.Harley, C. B., Vaziri, H., Counter, C. M., and
Allsopp, R. C. (1991) Exp. Gerontol., 27,
10114-10119.
5.Harley, C. B. (1991) Mutation Res.,
256, 271-282.
6.Harley, C. B., Futcher, A. B., and Greider, C. W.
(1990) Nature, 345, 458-460.
7.Allsopp, R. C., and Harley, C. B. (1995) Exp.
Cell Res., 219, 130-136.
8.Allsopp, R. C., Vaziri, H., Patterson, C.,
Goldstein, S., Younglai, E. V., Futcher, A. B., Greider, C. W., and
Harley, C. B. (1992) Proc. Natl. Acad. Sci. USA, 89,
10114-10118.
9.Vaziri, H., Dragowska, W., Allsopp, R. C., Thomas,
T. E., Harley, C. B., and Lansdorp, P. M. (1994) Proc. Natl. Acad.
Sci. USA, 91, 9857-9860.
10.Vaziri, H., Schachter, F., Uchida, I., Wei, L.,
Zhu, X., Effros, R., Cohen, D., and Harley, C. B. (1993) Am. J. Hum.
Genet., 52, 661-667.
11.Counter, C. M., Avilion, A. A., LeFeuvre, C. E.,
Stewart, N. G., Greider, C. W., Harley, C. B., and Bacchetti, S. (1992)
EMBO J., 11, 1921-1929.
12.Morin, G. B. (1989) Cell, 59,
521-529.
13.Kim, N. W., Piatyszek, M. A., Prowse, K. R.,
Harley, C. B., West, M. D., Ho, P. L., Coviello, G. M., Wright, W. E.,
Weinrich, S. L., and Shay, J. W. (1994) Science, 266,
2011-2015.
14.Bryan, T. M., Englezou, A., Gupta, J., Bacchetti,
S., and Reddel, R. R. (1995) EMBO J., 14, 4240-4248.
15.Vaziri, H., and Benchimol, S. (1996) Exp.
Gerontol., 31, 295-301.
16.El-Deiry, W. S., Tokino, T., Velculescu, V. E.,
Levy, D. B., Parson, R., Trent, J. M., Lin, D., Mercer, W. E., Kinzler,
K. W., and Vogelstein, B. (1993) Cell, 75, 817-825.
17.Harper, J. W., Adami, G. R., Wei, N., Keyomarsi,
K., and Elledge, S. J. (1993) Cell, 75, 805-816.
18.Di Leonardo, A., Linke, S. P., Clarkin, K., and
Wahl, G. M. (1994) Genes Dev., 8, 2540-2551.
19.Noda, A., Ning, Y., Venable, S. F.,
Pereira-Smith, O. M., and Smith, J. R. (1994) Exp. Cell Res.,
211, 90-98.
20.Shay, J. W., Wright, W. E., Brasiskyte, D., and
van der Haegen, B. A. (1993) Oncogene, 8, 1407-1413.
21.Bischoff, F. Z., Yim, S. O., Pathak, S., Grant,
G., Siciliano, M. J., Giovanella, B. C., Strong, L. C., and Tainsky, M.
A. (1990) Cancer Res., 50, 7979-7984.
22.Rogan, E. M., Bryan, T. M., Hukku, B., Maclean,
K., Chang, A. C., Moy, E. L., Englezou, A., Warneford, S. G.,
Dalla-Pozza, L., and Reddel, R. R. (1995) Mol. Cell. Biol.,
15, 4745-4753.
23.Kastan, M. B., Onyekwere, O., Sidransky, D.,
Vogelstein, B., and Craig, R. W. (1991) Cancer Res., 51,
6304-6311.
24.Bond, J. A., Wyllie, F. S., and Wynford-Thomas,
D. (1994) Oncogene, 9, 1885-1889.
25.Gollahon, L. S., and Shay, J. W. (1996)
Oncogene, 12, 715-725.
26.Irving, J., Geng, J., Wistrom, C., Pikaart, M.,
and Villeponteau, B. (1992) Exp. Cell. Res., 202,
61-66.
27.Atadja, P., Wong, H., Garkavtsev, I., Veillette,
C., and Riabowol, K. (1995) Proc. Natl. Acad. Sci. USA,
92, 8348-8352.
28.Kulju, K. S., and Lehman, J. M. (1995) Exp.
Cell. Res., 217, 336-345.
29.Vaziri, H., West, M. D., Allsopp, R. C., Davison,
T. S., Wu, Yu-Sung, Arrowsmith, C. H., Poirier, G. G., and Benchimol,
S. (1997) EMBO J., in press.
30.Benn, P. (1976) Amer. J. Hum. Genet.,
28, 465-473.
31.Michieli, P., Chedid, M., Lin, D., Pierce, J. H.,
Mercer, W. E., and Givol, D. (1994) Cancer Res., 54,
3391-3395.
32.Ogryzko, V. V., Schiltz, R. L., Russanova, V.,
Howard, B. H., and Nakatani, Y. (1996) Cell, 87,
3-9.
33.Shiloh, Y. (1995) Eur. J. Hum. Genet.,
3, 116-138.
34.Ambrose, H. J., Byrd, P. J., McConville, C. M.,
Cooper, P. R., Stankovic, T., Riley, J. H., Shiloh, Y., McNamara, J.
O., Fukao, T., and Taylor, A. M. (1994) Genomics, 21,
612-619.
35.Savitsky, K., Sfez, S., Tagle, D. A., Ziv, Y.,
Sartiel, A., Collins, F. S., Shiloh, Y., and Rotman, G. (1995) Hum.
Mol. Genet., 4, 2025-2032.
36.Greenwell, P. W., Kronmal, S. L., Porter, S. E.,
Gassenhuber, J., Obermaier, B., and Petes, T. D. (1995) Cell,
82, 823-829.
37.Brush, G. S., Morrow, D. M., Hieter, P., and
Kelly, T. J. (1996) Proc. Natl. Acad. Sci. USA, 93,
15075-15080.
38.Hari, K. L., Santerre, A., Sekelsky, J. J.,
McKim, K. S., Boyd, J. B., and Hawley, R. S. (1995) Cell,
82, 815-821.
39.Hartley, K. O., Gell, D., Smith, G. C., Zhang,
H., Divecha, N., Connelly, M. A., Admon, A., Lees-Miller, S. P.,
Anderson, C. W., and Jackson, S. P. (1995) Cell, 82,
849-856.
40.Shiloh, Y., Tabor, E., and Becker, Y. (1982)
Exp. Cell. Res., 140, 191-199.
41.Metcalfe, J. A., Parkhill, J., Campbell, L.,
Stacey, M., Biggs, P., Byrd, P. J., and Taylor, A. M. (1996) Nature
Genet., 13, 350-353.
42.Kastan, M. B., Zhan, Q., El-Deiry, W. S.,
Carrier, F., Jacks, T., Walsh, W. V., Plunkett, B. S., Vogelstein, B.,
and Fornace, A. J., Jr. (1992) Cell, 71, 587-590.
43.Zhang, N., Song, Q., Lu, H., and Lavin, M. F.
(1996) Oncogene, 13, 655-659.
44.Lu, X., and Lane, D. P. (1993) Cell,
75, 765-778.