Telomerase: a Promising Marker of Biological Immortality of Germ, Stem, and Cancer Cells. A Review
A. K. Meeker1 and D. S. Coffey1,2
1James Buchanan Brady Urological Institute Research Laboratories, The Johns Hopkins Hospital, Baltimore, Maryland 21287-2101, USA; fax: (410) 955-0833; E-mail: DCoffey@gwgate1.welch.jhu.edu2To whom correspondence should be addressed.
Submitted September 12, 1997.
This review will describe the current state of knowledge of telomerase as it relates to human malignancies, focusing primarily on published measurements of this enzyme’s activity in benign and malignant neoplasms and their normal tissue counterparts. Key questions concerning the potential clinical utility of assaying for telomerase activity will be addressed and the implications of recent findings discussed.
KEY WORDS: telomerase, oncogenesis, cancer.
In the 1960’s, Leonard Hayflick and Paul Moorhead overturned what was then a reigning paradigm in cell biology--that under optimal growth conditions, cells possessed an unlimited capacity for replication. Their elegant series of experiments convincingly showed that, on the contrary, human cells displayed a strictly limited division potential [1, 2]. Thus, the currently prevailing view holds that dividing human cells, both in vitro and in vivo, eventually age and senesce. However, as with most hypotheses in biology, informative exceptions exist including: germline cells, "immortalized" tissue culture cell lines, and cancer cells. These three classes of cells have somehow overcome the limitations of cellular replication and aging. Thus, for example, aged parents always give birth to offspring containing young cells. It seems reasonable to assume then, that characteristics unique to these three types of cells are likely to be fundamental pre-requisites for unlimited growth. One such characteristic these cell types share is activity of the enzyme telomerase. Understanding the mechanism and regulation of this enzyme could therefore provide insight into the processes of aging, immortalization, and cancer.
Background: Telomeres and Telomerase
In 1971, following the elucidation of the mechanism of DNA replication, Alexey Olovnikov pointed out the existence of what has since been called the end replication problem; that is, the incomplete duplication by DNA polymerase of the 5´ ends of linear DNA molecules [3]. Without a compensatory mechanism, this loss of terminal DNA sequences should result in the shortening of chromosomes with each round of cell division. Twenty five years ago, Dr. Olovnikov outlined his "Theory of Marginotomy" in which he proposed that this progressive chromosome shortening might underlie the limited division potential observed in normal somatic cells grown in vitro [1, 2]. It was in turn proposed that this limited growth potential could be an underlying cause of organismal aging and age-related disorders. In addition, Olovnikov suggested that malignant cells must possess some mechanism for circumventing the end replication problem [4].
How cells could withstand such progressive chromosome shortening at all was explained when the structure of the chromosomal termini, the telomeres, became known. Telomeres are specialized structures at the chromosome ends of nearly all eukaryotes, composed of repetitive short DNA sequences and associated telomeric binding proteins (reviewed in [5]). Since telomeric sequences are non-coding they serve as buffer zones against the telomeric attrition caused by the end-replication problem. For example, the chromosomes of human somatic cells are capped by roughly 10 kilobase pairs of the repeated hexamer sequence TTAGGG. Approximately 50-100 bp of this telomeric sequence is lost with each cell division, thus such a cell could theoretically withstand at least 100 divisions before any loss of coding sequences proximal to the telomeres occurred. However, some chromosomes are likely to have telomeres much shorter than the mean value. Furthermore, telomeres appear to provide a protective capping function, preventing degradation as well as undesirable recombination events such as chromosome end-to-end fusions commonly associated with broken chromosome ends and these functions may be compromised well before total loss of the telomeric sequence occurs. The loss of either of these functions may be the limiting factor that blocks continued cell division.
The predicted shortening of linear chromosomes in somatic tissues was borne out by several studies which indicated that such shortening indeed occurred as a function of donor age for primary tissue samples, and duration of in vitro growth in the case of explanted tissue cultures [6-9]. With this new knowledge of telomere structure and dynamics, Olovnikov’s marginotomy theory has since been restated as the "telomere hypothesis of cellular aging" ([10], reviewed in [11]). Aging is a complicated process, but telomere loss is one critical step.
But what about single celled eukaryotes? Populations of these organisms have essentially unlimited replication capacities and, therefore, must posses a mechanism for overcoming the end-replication problem. In 1985, Greider and Blackburn discovered an enzyme in Tetrahymena thermophila which was able to replace the lost telomeric sequences, thereby maintaining chromosomal integrity [12]. This enzyme, which they named telomerase, was shown to contain an essential RNA component, a portion of which serves as the template for the synthesis of the telomeric sequence, thus placing telomerase within the family of reverse transcriptases [13, 14].
In 1989, telomerase activity was detected by Morin in an immortalized human cancer cell line (HeLa), and later by Counter during the in vitro immortalization of human embryonic kidney cells whose telomere lengths were then seen to stabilize [15, 16]. Finally, in 1994, telomerase activity was detected in human ovarian and B-cell cancers, confirming Olovnikov’s prediction of an "anti-marginotomy" mechanism at work in malignant cells [17, 18]. Unfortunately, the primer extension assay used up to this point for detecting telomerase activity required large amounts of tissue, making widespread application of the method difficult. Then, in December of 1994, Kim et al. published their Telomeric Repeat Amplification Protocol (TRAP) which featured an improved tissue lysis method and PCR amplification of the telomerase-extended oligonucleotide substrate [19]. These improvements resulted in a 10,000-fold increase in assay sensitivity, thereby allowing the use of very small (milligram or less) amounts of tissue. Since then, there has been a flurry of investigations in which telomerase activity has been assayed in normal, benign lesions, pre-malignant, and cancerous samples of many human tissue types, using either the original or one of various modified versions of the TRAP assay as there is no currently agreed upon standard method for measuring or quantitating activity.
Surveys of Telomerase Activity in Human Tumors
As shown in Table 1, more than 2600 human tumor samples have been tested for telomerase activity using the PCR-based TRAP assay. The overall prevalence of 84.9% makes telomerase activity the most common biochemical marker of human cancer.
TABLE 1. Survey Results of Telomerase
Activity in Human Malignanciesa
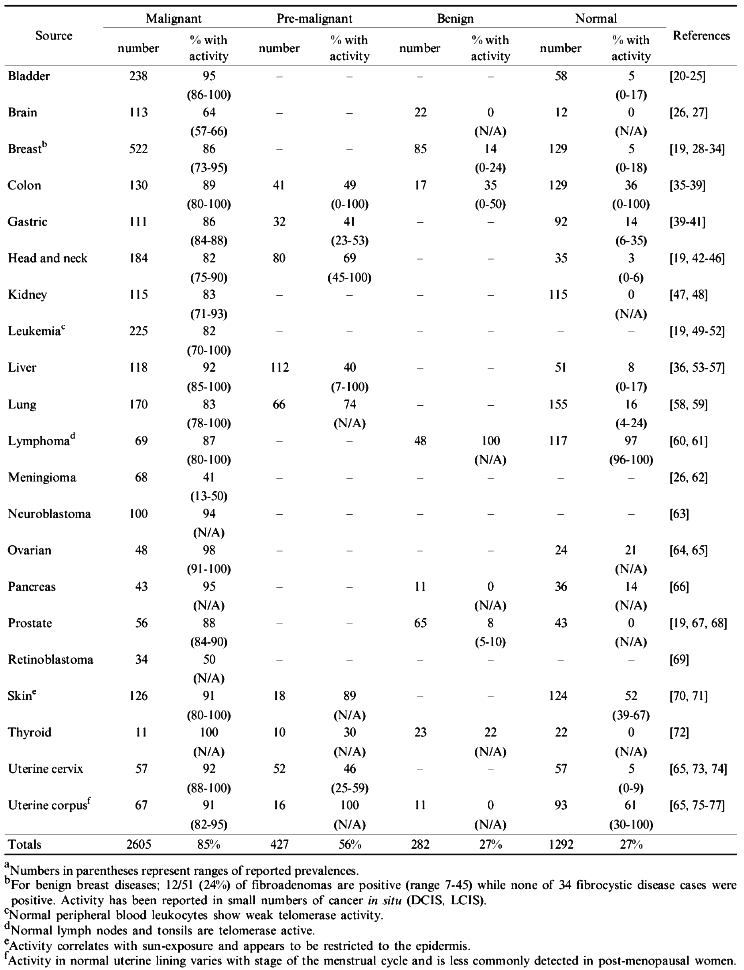
The fact that telomerase is frequently activated in the majority of tumor types suggests that information on telomerase activity status may have clinical utility in cancer screening, diagnosis, and monitoring treatment efficacy. To evaluate these possibilities, the following questions should be addressed.
1. To what extent is telomerase activity specific for the malignant state as opposed to pre-malignant, benign, or normal tissue counterparts?
2. When in the multi-step process of tumorigenesis is telomerase activated?
3. How sensitive and robust are current assays of telomerase activity?
4. What are the proper criteria and controls necessary to confidently classify a sample as either telomerase positive or negative?
5. Can telomerase activity be reliably quantified and is such quantitative information useful?
6. How does telomerase compare with other "gold standard" tumor markers?
Specificity of Telomerase Activity in Cancer
In order for the measurement of telomerase activity to be useful clinically, either its presence must be restricted to malignant tissues or activity levels must be quantitatively distinguishable from those present in normal and benign tissues. Early surveys of telomerase activity in human material found only rare instances of active telomerase in non-malignant tissues compared to the vast majority of cancers which were discovered to be telomerase positive (reviewed in [78]). These initial observations supported the hypothesis that cancer cells were primarily telomerase-positive and immortal while their normal somatic cell counterparts were telomerase-negative and mortal. This is in keeping with the observed telomere shortening and limited life spans of non-neoplastic cells. Since then however, the widespread use of sensitive PCR-based assays has resulted in the detection of activity in increasing numbers of some types of both normal and benign tissues.
The fact that some normal tissues possess telomerase activity was not unanticipated. Within our own research group we hypothesized that the stem cell pools of regenerative tissues such as the hematopoietic system, the lining of the gut, and the epidermis of the skin, would likely require active telomerase in order to sustain the large amount of cell turnover observed in such systems [79]. We tested this hypothesis in male rodent secondary sex-accessory tissues which can be experimentally cycled to grow, involute, and regrow by manipulation of serum testosterone levels. In the prostate for example, lowering testosterone causes glandular involution and the loss of over 90% of prostate cells. The small residual prostate and seminal vesicles that remain following testosterone withdrawal are highly enriched for stem cells and display high levels of telomerase activity. Furthermore, activity is rapidly down-regulated in conjunction with glandular regeneration which occurs following the re-introduction of testosterone [80].
The first indication that normal human tissues possessed active telomerase came from the discovery that peripheral blood leukocytes and bone marrow cells from normal donors had detectable levels of enzyme activity [49, 81, 82]. This was surprising because it was known from previous studies that these same cell populations exhibited telomere shortening. Interestingly, in the hematopoietic system, telomerase activity was not restricted to the most primitive stem cells but was also found in mature T-cells, B-cells, macrophages, and granulocytes [49, 82, 83]. Although present, the activity detected was generally much weaker than that found in established lines of immortal tissue culture cells or in malignant cells. However, in vitro studies have shown that activity can be up-regulated by a number of activating mitogenic stimuli including: phytohemagglutinin in T-cells, pokeweed mitogen in B-cells, phorbol ester/Ca-ionophore combinations, and various anti-cell surface receptor antibodies [82-90].
In addition to bone marrow and peripheral blood cells, normal and benign lymph nodes have also been shown to be telomerase-positive, with activity localized to proliferating B-cells in the germinal centers of secondary follicles [59, 91]. Moreover, recent studies have shown that activity clearly exists in normal skin and cervical epidermis [70, 71, 92, 93], hair follicles [71, 94], the endometrium of the uterus [75-77], vascular endothelial cells [95], as well as the lower portions of the crypts within the colonic mucosa [38]. All of these tissues are undergoing continuous cell turnover and, where examined, telomerase activity appears restricted to regions harboring stem cells and proliferating amplifying cell pools, while this activity is apparently lost in other zones in conjunction with terminal differentiation.
As indicated in Table 1, the overall prevalence of telomerase activity in normal and benign neoplastic solid tissues are both 27%, significantly greater than in earlier reports. However, lymph nodes, which are nearly always found to be positive, represent greater than one-half of the benign and approximately one-third of the normal positive samples making up this average. The bulk of the remaining positive cases come from tissues with renewal and regenerative properties such as the skin, uterus, and colon. If these three sources, along with lymph nodes, are excluded from consideration in the overall averages then the incidence of telomerase-positive cells in normal solid tissues drops to 6%. On the whole it seems that, in addition to the aforementioned telomerase activity found in normal blood and bone marrow, activity is also commonly detected in non-cancerous samples of other actively cycling tissues. This background activity may compromise the clinical value of telomerase assays in these regenerating tissue types. If activity could be reliably quantitated it might be possible to set reasonable cut off limits that would differentiate between cancerous and non-cancerous samples. Indeed, where it has been assessed, activity levels are generally far lower in normal and benign tissues than in their malignant counterparts. However, as has been shown for leukemias, there may be significant overlap in activity levels between cancer and non-cancer samples [49].
Lastly, it should be pointed out that false positive signals are of some concern. In many instances the normal and/or benign tissue samples assayed are obtained from the same clinical specimens as are the tumor samples. This means that contamination of the normal/benign sample with tumor cells could occur, either during sample processing, or within the sample itself due to existing metastatic spread of the tumor cells. Also, it is unclear what contribution lymphocytes, especially activated ones, may make to the telomerase activity detected in a given sample. Studies have indicated that, in order to produce a positive signal in the TRAP assay, at least 103-104 peripheral blood leukocytes are required [51, 58, 82, 86, 96]. However, as mentioned previously, activity is significantly up-regulated during leukocyte activation. Indeed, cases of telomerase activity apparently derived from such cells has been reported for a number of inflammatory conditions such as contact dermatitis [70], cirrhosis of the liver [53, 55-57], and thyroid adenomas [72].
When is Telomerase Activated during Cancer Development?
Telomerase activity has been detected in both germline cells and in the developing embryo [19, 63, 91, 97]. However, activity is turned off in most somatic cells of the neonate although, as discussed in the previous section, low levels of activity seem to persist in regenerative tissues. Telomerase is found to be reactivated or upregulated in the majority of cancers. If this activation occurs early during tumorigenesis, then it may be a useful marker for cancer detection and early diagnosis. If, on the other hand, it is a late event, then it may be better suited for staging or prognostic purposes.
Specific cancers in which there is evidence that activation occurs early include the following: bladder [20, 22, 25], liver [53, 54, 56], colorectal [39], head and neck [42, 43], lung [59], cervix [73, 74], kidney [47, 48], prostate [67], and thyroid [72]. In these tumor types, neither the presence or absence of activity, nor the levels of activity correlate with markers known to have prognostic value.
Cancers in which there does appear to be a correlation between activity status and tumor grade, stage, prognostic factors, or patient survival, include: meningiomas [26, 62], neuroblastomas [63], non-Hodgkins lymphomas [61], and certain leukemias [50, 51].
Conflicting results have been reported for gastric and breast cancers. In the breast, activity has been found in pre-malignant lesions and most studies fail to find any link between activity and known prognostic factors [30-33]. However, in a large series, Hiyama et al., reported a statistically significant correlation between lack of activity and early tumor stage [28]. For gastric cancers, Hiyama et al., found that activity correlated with tumor size, presence of metastasis to lymph nodes, and survival [40], yet this was not seen in the study by Tahara et al., which examined a smaller number of patients [39].
Sensitivity and Other Assay Characteristics
In conducting the TRAP assay, cells or tissues are extracted with a detergent-based lysis buffer. An oligonucleotide substrate is then added to the cleared lysate and if active telomerase is present, this primer will be extended by the addition of telomeric sequences by the telomerase holoenzyme. This extended primer is then amplified via PCR, and the products are detected by one of a variety of methods. The assay is exquisitely sensitive, able to detect activity even in single cells under optimized conditions [98]. This sensitivity allows for the use of very small tissue samples and should facilitate use in cancer screening; either in the general population or in subgroups deemed to be at risk for developing a particular malignancy. It must be pointed out, however, that assaying telomerase activity is subject to many pitfalls (Table 2) requiring rigorous attention to detail, use of proper positive and negative controls, and care in interpretation of the results. It is important to note that many published studies have not made careful use of such controls; this should be borne in mind when considering the conclusions presented in such studies.
TABLE 2. Potential Problems in Assaying
Telomerase Activity

One concern with current assays is that false negative results may arise from a number of causes, and therefore, proper controls must be included in order to insure that the results provide meaningful information. One potential problem is that telomerase could be inactivated or inhibited in the assay. Telomerase could be inactivated due to inadequate handling or preservation of tissue samples and cell extracts, by endogenous proteases, and by endogenous or contaminating RNAses which result in hydrolysis of the telomerase RNA subunit. The overall integrity of a cell lysate can be checked by assaying for other enzyme activities such as alkaline phosphatase or DNA polymerase, and the presence of hydrolytic enzymes can be checked by the addition of RNA or protein standards which can then be examined for evidence of their degradation. Also, inclusion of an extract known to be telomerase-positive tells whether or not the assay is functioning as expected. An additional worry is that many tissues contain inhibitors of the PCR portion of the TRAP assay [38, 44, 96, 98]. Therefore, an important positive control, which tests for tissue-derived assay inhibitors, is to dope each cell extract with a PCR-amplifiable DNA standard that can be identified as a specific fluorescence peak or band on a gel [98, 99]. Detection of the amplification product of such a standard provides information on testing the operation of the PCR arm of the assay and indicates that a lack of telomerase signal is not due to PCR-inhibition. Two other methods used to check for inhibitors are the mixing of negative extracts with known positive ones and assaying several dilutions of the cell lysate in the hopes of diluting out any inhibitors (although low levels of telomerase activity may also be diluted out).
Tissue sampling artifacts are another potential source of false negatives in cancer studies. Regions lacking telomerase may exist within a sample due to heterogeneity of enzyme expression, areas of necrosis, or even a lack of tumor cells in the small sample sizes typically processed for testing. Notably, heterogeneous expression of both the telomerase RNA component, as well as enzymatic activity have been observed [33, 100]. These potential problems underscore the importance of careful histologic examination of the tissue sample wherever possible. In addition, sampling artifacts are also likely causes of the reduced sensitivity of telomerase detection seen in several studies in which very small samples are obtained; for example, by isolating cells directly from the urine or form saline rinses of the bladder, oral cavity, or colon, as opposed to direct sampling of surgically removed tissue. Reduced sensitivities have also been observed when using cells from needle aspirates as well as exfoliated cells from tissue biopsies.
A second concern with current assays is the problem of false positives. These may arise due to PCR product contamination, cross-contamination from active tissues, synthetic activities other than telomerase present in the lysate, or positive signals arising from extensions of dimerized PCR primers in the reaction mix. A genuine telomerase signal is primer-dependent, and should be abolished in control lysates pre-treated with either protease, RNAse, or heat. Avoidance of PCR-product contamination is important, especially since the same telomeric sequence is being amplified in each reaction. For this reason, reactions without added cell extract are necessary negative controls. An additional worrisome problem is the fact that the PCR primers can associate with each other and then be extended by Taq polymerase. Subsequent rounds of staggered annealing and amplification can produce PCR products indistinguishable from bona fide telomerase-derived products. Telomerase activity produces a discrete ladder of bands with a six base pair periodicity in the TRAP assay when the products are visualized by autoradiography or fluorescence following gel electrophoresis. Primer dimers are readily extended to produce the first few of these bands (discussed in [101]). This problem has been lessened by the use of modified primer sequences that discourage spurious extension [99, 101]. However, overestimation of telomerase prevalence in studies using the original primer sequences remains a potential problem.
Finally, the sensitivity of the method chosen for detection of PCR products can affect one’s conclusions. For example, in autoradiography, a sample initially thought to lack telomerase activity may be revealed as telomerase-positive upon longer exposure.
Quantitation of Telomerase Activity
Several attempts have been made at quantifying telomerase activity; ranging from subjective ratings of assay signal intensities to computerized densitometric analyses that control for background noise and assay inhibitors. Perhaps the most reliable current method is the latest modified version of the original TRAP assay protocol [99]. Quantitative results are probably most meaningfully expressed on a per cell basis, but this is often difficult or impossible to do when using clinical tissue samples; therefore, results are often presented on a per protein basis. Even on a per cell basis however, results may not be informative unless one also knows the histology of the tissue sample being analyzed. For example, tumor metastases often have higher levels of telomerase activity than those found in the original tumor. The primary tumor is however, often much more heterogeneous; including normal epithelial and stromal cells, as well as varying amounts of extracellular matrix and connective tissue, all of which may act to decrease the proportion of cancer cells in the sample.
Telomerase and Tumor Biology
The prevalence of telomerase activity in human cancers indicates that telomeric stabilization is likely to be an important factor in tumorigenesis and implies that tumors are made up, at least in part, of immortalized cells. How it is that tumor cells acquire this activity remains an open question although two hypotheses have been proposed [81]. The reactivation model postulates that cancer is initiated in a telomerase-negative cell which then reactivates telomerase at some point during tumorigenesis in order to halt the telomeric shortening that would otherwise block the multiple clonal expansions required to develop malignancy. Alternatively, the retention model proposes that cancer develops in a cell in which telomerase is already active and this activity is subsequently retained or even up-regulated during tumorigenesis.
But, what is to be made of the 15% of human tumors that are apparently telomerase-negative? Are these cells still subject to the Hayflick limit and therefore mortal? Or, are other, telomerase-independent mechanisms for telomeric stabilization available to these cells? Non-telomerase pathways for telomere maintenance have been documented in other organisms. Yeast, for example, are able to maintain their telomeres by genetic recombination following experimental inactivation of telomerase. Examples have been found of immortalized human cell lines that apparently lack telomerase activity [19, 102-105]. However, there are currently very few examples of such cell lines, and these were derived during in vitro immortalization studies rather than established from human tumors. In addition, such cells exhibit a very long and heterogeneous distribution of telomere lengths when they are analyzed by Southern blotting techniques; a pattern which has not been reported for any telomerase-negative human tumor [19, 106]. To date, the literature seems to indicate that if tumor cells can make use of telomerase-independent mechanisms to stabilize their telomeres it is a rare event. Interestingly, the introduction into HeLa cells of an antisense construct targeted against the telomerase RNA component resulted in telomere shortening followed by cell death indicating that, at least in this human, tumor-derived immortal cell line, alternate mechanisms were not activated [107].
The means by which telomerase activity is regulated is a topic currently receiving much attention. In general, one can identify three normal modes of telomerase regulation in human tissues. First, there is an apparently irreversible loss of activity in certain somatic tissues during development, for example, in terminally differentiated muscle or nerve cells. Second, activity is preserved in the germline in order to maintain the lengths of the chromosomes which will be passed on to future generations. Third, regenerative tissues, such as the hematopoietic system and epithelial coverings and linings, retain telomerase activity in order to support the large amounts of cell turnover occurring in these tissues. Activity in renewal tissues generally, though not exclusively, correlates with cell proliferation and may be subject to hormonal regulation as is the case for the uterus and prostate.
How telomerase becomes dysregulated in cancer cells remains an important unanswered question. Studies indicate that some tumor-derived cell lines retain the ability to down-regulate activity upon induction of differentiation while other cell lines do not show this. Comparisons of the levels of the telomerase RNA component with enzyme activity generally seem to indicate that such RNA levels are not the limiting factor in regulating enzymatic activity [108, 109].
Recently, the catalytic subunit of human telomerase, located on chromosome 5, has been cloned and initial results support the view that activity is regulated at the level of the protein [110, 111].
Summary and New Directions
The end replication problem sets a finite barrier to cell division and must be overcome in established cell lines and tumors. The evidence to date indicates that the vast majority of immortal human cells accomplish this through the use of the enzyme telomerase. The presence of tightly regulated or low levels of telomerase activity in normal regenerative tissues probably reflects their large replicative potentials, allowing for continuous cell renewal in such tissues by slowing telomeric loss, but not preventing it altogether.
Telomerase is currently the best and most common general marker of cancer cells. Activity has been detected in every major category of human malignant neoplasia tested, exhibiting an overall prevalence of 85%. Accordingly, intense interest in the use of this marker for the purposes of diagnosis and/or prognosis has been generated in the cancer research community. This field is still relatively young and further work is needed to improve and standardize current methods for detecting and quantitating telomerase activity. Also needed are more extensive clinical studies incorporating larger numbers of patients and longer periods of patient follow-up to help clarify telomerase’s potential diagnostic and prognostic roles in the various types of human cancer.
The cloning of the genes for the RNA component [107] and, more recently, the catalytic protein subunit [110, 111] of human telomerase opens up the prospects of developing new in situ assays for the detection and detailed localization of these critical enzyme components. These new tools will provide information on the important issues of the tissue localization and regulation of this enzyme.
The RNA subunit has already been used as a target for testing antisense [107], ribozyme [112], and peptide nucleic acid [113] strategies aimed at inhibiting telomerase activity with an eye towards therapeutic applications. Characterization of the newly cloned protein subunits as well as illumination of the enzymatic mechanism of telomerase will hopefully provide useful new targets for our continuing war on cancer.
This work was supported by NCI spore Grant NCICA58236 and NIH Training Grant T32GMo7445-18. The authors wish to thank Dan Krovich and Angelo DeMarzo for critical reading of the manuscript and helpful discussions.
LITERATURE CITED
1.Hayflick, L. (1965) Exp. Cell Res.,
37, 614-636.
2.Hayflick, L., and Moorhead, P. S. (1961) Exp.
Cell Res., 25, 585-621.
3.Olovnikov, A. M. (1971) Dokl. Akad. Nauk
SSSR, 201, 1496-1499.
4.Olovnikov, A. M. (1973) J. Theor. Biol.,
41, 181-190.
5.Blackburn, E. H. (1991) Nature, 350,
569-572.
6.Hastie, N., Dempster, M., Dunlop, M., Thompson, A.,
Green, D., and Alshire, R. (1990) Nature, 346,
866-868.
7.Lindsey, J., McGill, N. I., Lindsey, L. A., Green,
D. K., and Cooke, H. J. (1991) Mutat. Res., 256,
45-48.
8.Harley, C. B., Futcher, A. B., and Greider, C. W.
(1990) Nature, 345, 458-460.
9.Allsopp, R. C., Vaziri, H., Patterson, C.,
Goldstein, S., Younglai, E. V., Futcher, A. B., Greider, C. W., and
Harley, C. B. (1992) Proc. Natl. Acad. Sci. USA, 89,
10114-10118.
10.Harley, C. B., Vaziri, H., Counter, C. M., and
Allsopp, R. C. (1992) Exp. Gerontol., 27, 375-382.
11.Holt, S. E., Shay, J. W., and Wright, W. E.
(1996) Nature Biotechnol., 14, 1-4.
12.Greider, C. W., and Blackburn, E. H. (1985)
Cell, 43, 405-413.
13.Greider, C. W., and Blackburn, E. H. (1989)
Nature, 337, 331-336.
14.Greider, C. W., and Blackburn, E. H. (1987)
Cell, 51, 887-898.
15.Morin, G. (1989) Cell, 59,
521-529.
16.Counter, C., Avilion, A., LeFeuvre, C., Stewart,
N., Greider, C., Harley, C., and Bacchetti, S. (1992) EMBO J.,
11, 1921-1929.
17.Counter, C. M., Hirte, H. W., Bacchetti, S., and
Harley, C. B. (1994) Proc. Natl. Acad. Sci. USA, 91,
2900-2904.
18.Nilsson, P., Mehle, C., Remes, K., and Roos, G.
(1994) Oncogene, 9, 3043-3048.
19.Kim, N. W., Piatyszek, M. A., Prowse, K. R.,
Harley, C. B., West, M. D., Ho, P. L., Coviello, G. M., Wright, W. E.,
Weinrich, S. L., and Shay, J. W. (1994) Science, 266,
2011-2015.
20.Muller, M., Heine, B., Heicappel, R., Emrich, T.,
Hummel, M., Stein, H., and Miller, K. (1996) Int. J. Oncol.,
9, 1169-1173.
21.Lin, Y., Miyaoto, H., Fujinami, K., Uemura, H.,
Hosaka, M., Iwasaki, Y., and Kubota, Y. (1996) Clin. Cancer
Res., 2, 929-932.
22.Yoshida, K., Sugino, T., Tahara, H., Woodman, A.,
Bolodeoku, J., Nargund, V., Fellows, G., Goodison, S., Tahara, E., and
Tarin, D. (1997) Cancer, 79, 362-369.
23.Kamata, S., Kageyama, Y., Yonese, J., and Oshima,
H. (1996) Br. J. Urol., 78, 704-708.
24.Kinoshita, H., Ogawa, O., Kakehi, Y., Mishina,
M., Mitsumori, K., Itoh, N., Yamada, H., Terachi, T., and Yoshida, O.
(1997) J. Natl. Cancer Inst., 89, 724-730.
25.Kyo, S., Kunimi, K., Uchibayashi, T., Namiki, M.,
and Inoue, M. (1997) Am. J. Clin. Pathol., 107,
555-560.
26.Demasters, B. K. K., Markham, N., Lillehei, K.
O., and Shroyer, K. R. (1997) Am. J. Clin. Pathol., 107,
548-554.
27.Langford, L. A., Piatyszek, M. A., Xu, R.,
Schold, S. C., Jr., and Shay, J. W. (1995) Lancet, 346,
1267-1268.
28.Hiyama, E., Gollahon, L., Kataoka, T., Kuroi, K.,
Yokoyama, T., Gazdar, A. F., Hiyama, K., Piatyszek, M. A., and Shay, J.
W. (1996) J. Natl. Cancer Inst., 88, 116-122.
29.Mokbel, K., and Ghilchik, M. (1996) J. Natl.
Cancer Inst., 88, 839-840.
30.Sugino, T., Yoshida, K., Bolodeoku, J., Tahara,
H., Buley, I., Manek, S., Wells, C., Goodison, S., Ide, T., Suzuki, T.,
Tahara, E., and Tarin, D. (1996) Int. J. Cancer, 69,
301-306.
31.Bednarek, A. K., Sahin, A., Brenner, A. J.,
Johnston, D. A., and Aldaz, C. M. (1997) Clin. Cancer Res.,
3, 11-16.
32.Nawaz, S., Hashizumi, T. L., Markham, N. E.,
Shroyer, A. L., and Shroyer, K. R. (1997) Am. J. Clin. Pathol.,
107, 542-547.
33.Tsao, J. I., Zhao, Y. L., Lukas, J., Yang, X. W.,
Shah, A., Press, M., and Shibata, D. (1997) Clin. Cancer Res.,
3, 627-631.
34.Landberg, G., Nielsen, N. H., Nilsson, P., Emdin,
S. O., Cajander, J., and Roos, G. (1997) Cancer Res.,
57, 549-554.
35.Li, Z. H., Salovaara, R., Aaltonen, L. A., and
Shibata, D. (1996) Am. J. Pathol., 148, 1075-1079.
36.Chadeneau, C., Hay, K., Hirte, H. W., Gallinger,
S., and Bacchetti, S. (1995) Cancer Res., 55,
2533-2536.
37.Yoshida, K., Sugino, T., Goodison, S., Warren, B.
F., Nolan, D., Wadsworth, S., Mortensen, N. J., Toge, T., Tahara, E.,
and Tarin, D. (1997) Br. J. Cancer, 75, 548-553.
38.Hiyama, E., Hiyama, K., Tatumoto, N., Kodama, T.,
Shay, J., and Yokoyama, T. (1996) Int. J. Oncol., 9,
453-458.
39.Tahara, H., Kuniyasu, H., Yokozaki, H., Yasui,
W., Shay, J., Ide, T., and Tahara, E. (1995) Clin. Cancer Res.,
1, 1245-1251.
40.Hiyama, E., Yokoyama, T., Tatsumoto, N., Hiyama,
K., Imamura, Y., Murakami, Y., Kodama, T., Piatyszek, M. A., Shay, J.
W., and Matsuura, Y. (1995) Cancer Res., 55,
3258-3262.
41.Kuniyasu, H., Domen, T., Hamamoto, T., Yokozaki,
H., Yasui, W., Tahara, H., and Tahara, E. (1997) Jap. J. Cancer
Res., 88, 103-107.
42.Mutirangura, A., Supiyaphun, P., Trirekapan, S.,
Sriuranpong, V., Sakuntabhai, A., Yenrudi, S., and Voravud, N. (1996)
Cancer Res., 56, 3530-3533.
43.Mao, L., Elnaggar, A. K., Fan, Y. H., Lee, J. S.,
Lippman, S. M., Kayser, S., Lotan, R., and Hong, W. K. (1996) Cancer
Res., 56, 5600-5604.
44.Califano, J., Ahrendt, S. A., Meininger, G.,
Westra, W. H., Koch, W. M., and Sidransky, D. (1996) Cancer
Res., 56, 5720-5722.
45.Hohaus, S., Cavallo, S., Bellacosa, A., Genuardi,
M., Galli, J., Cadoni, G., Almadori, G., Lauriola, L., Litwin, S.,
Maurizi, M., and Neri, G. (1996) Clin. Cancer Res., 2,
1895-1900.
46.Kannan, S., Tahara, H., Yokozaki, H., Mathew, B.,
Nalinakumari, K. R., Nair, K., and Tahara, E. (1997) Cancer,
Epidemiology, Biomarkers and Prevention, 6, 413-420.
47.Fiedler, W., Dahse, R., Schlichter, A., Junker,
K., Kosmehl, H., Ernst, G., Schubert, J., and Claussen, U. (1996)
Int. J. Oncol., 9, 1227-1232.
48.Mehle, C., Piatyszek, M. A., Ljungberg, B., Shay,
J. W., and Roos, G. (1996) Oncogene, 13, 161-166.
49.Broccoli, D., Young, J. W., and de Lange, T.
(1995) Proc. Natl. Acad. Sci. USA, 92, 9082-9086.
50.Ohyashiki, K., Ohyashiki, J. H., Iwama, H.,
Hayashi, S., Shay, J. W., and Toyama, K. (1997) Leukemia,
11, 190-194.
51.Ohyashiki, J. H., Ohyashiki, K., Iwama, H.,
Hayashi, S., Toyama, K., and Shay, J. W. (1997) Clin. Cancer
Res., 3, 619-625.
52.Zhang, W., Piatyszek, M., Kobayashi, T., Estey,
E., Andreeff, M., Deisseroth, A., Wright, W., and Shay, J. (1996)
Clin. Cancer Res., 2, 799-803.
53.Tahara, H., Nakanishi, T., Kitamoto, M.,
Nakashio, R., Shay, J. W., Tahara, E., Kajiyama, G., and Ide, T. (1995)
Cancer Res., 55, 2734-2736.
54.Ohta, K., Kanamaru, T., Yamamoto, M., and Saitoh,
Y. (1996) Kobe J. Med. Sci., 42, 207-217.
55.Nakashio, R., Kitamoto, M., Tahara, H.,
Nakanishi, T., Ide, T., and Kajiyama, G. (1997) Int. J. Cancer,
74, 141-147.
56.Kojima, H., Yokosuka, O., Imazeki, F., Saisho,
H., and Omata, M. (1997) Gastroenterology, 112,
493-500.
57.Miura, N., Horikawa, I., Nishimoto, A., Ohmura,
H., Ito, H., Hirohashi, S., Shay, J. W., and Oshimura, M. (1997)
Cancer Genet. Cytogenet., 93, 56-62.
58.Hiyama, K., Hiyama, E., Ishioka, S., Yamakido,
M., Inai, K., Gazdar, A. F., Piatyszek, M. A., and Shay, J. W. (1995)
J. Natl. Cancer Inst., 87, 895-902.
59.Yashima, K., Litzky, L., Kaiser, L., Rogers, T.,
Lam, S., Wistuba, I., Milchgrub, S., Srivastava, S., Piatyszek, M.,
Shay, J., and Gazdar, A. (1997) Cancer Res., 57,
2373-2377.
60.Brousset, P., Alsaati, T., Chaouche, N., Zenou,
R. C., Schlaifer, D., Chittal, S., and Delsol, G. (1997) Blood,
89, 26-31.
61.Norrback, K. F., Dahlenborg, K., Carlsson, R.,
and Roos, G. (1996) Blood, 88, 222-229.
62.Langford, L. A., Piatyszek, M. A., Xu, R. S.,
Schold, S. C., Wright, W. E., and Shay, J. W. (1997) Hum.
Pathol., 28, 416-420.
63.Hiyama, E., Hiyama, K., Yokoyama, T., Matsuura,
Y., Piatyszek, M. A., and Shay, J. W. (1995) Nature Med.,
1, 249-255.
64.Wan, M., Li, W. Z., Duggan, B. D., Felix, J. C.,
Zhao, Y., and Dubeau, L. (1997) J. Natl. Cancer Inst.,
89, 437-441.
65.Zheng, P. S., Iwasaka, T., Yamasaki, F., Ouchida,
M., Yokoyama, M., Nakao, Y., Fukuda, K., Matsuyama, T., and Sugimori,
H. (1997) Gynecol. Oncol., 64, 171-175.
66.Hiyama, E., Kodama, T., Shinbara, K., Iwao, T.,
Itoh, M., Hiyama, K., Shay, J. W., Matsuura, Y., and Yokoyama, T.
(1997) Cancer Res., 57, 326-331.
67.Sommerfeld, H. J., Meeker, A. K., Piatyszek, M.
A., Bova, G. S., Shay, J. W., and Coffey, D. S. (1996) Cancer
Res., 56, 218-222.
68.Lin, Y., Uemura, H., Fujinami, K., Hosaka, M.,
Harada, M., and Kubota, Y. (1997) J. Urol., 157,
1161-1165.
69.Gupta, J., Han, L. P., Wang, P., Gallie, B. L.,
and Bacchetti, S. (1996) J. Natl. Cancer Inst., 88,
1152-1157.
70.Taylor, R. S., Ramirez, R. D., Ogoshi, M.,
Chaffins, M., Piatyszek, M. A., and Shay, J. W. (1996) J. Invest.
Dermatol., 106, 759-765.
71.Ueda, M., Ouhtit, A., Bito, T., Nakazawa, K.,
Lubbe, J., Ichihashi, M., Yamasaki, H., and Nakazawa, H. (1997)
Cancer Res., 57, 370-374.
72.Umbricht, C. B., Saji, M., Westra, W. H.,
Udelsman, R., Zeiger, M. A., and Sukumar, S. (1997) Cancer Res.,
57, 2144-2147.
73.Pao, C. C., Tseng, C. J., Lin, C. Y., Yang, F.
P., Hor, J. J., Yao, D. S., and Hsueh, S. (1997) J. Clin.
Oncol., 15, 1932-1937.
74.Kyo, S., Takakura, M., Ishikawa, H., Sasagawa,
T., Satake, S., Tateno, M., and Inoue, M. (1997) Cancer Res.,
57, 1863-1867.
75.Brien, T., Kallakury, B., Lowry, C., Ambros, R.,
Muraca, P., Malfetano, J., and Ross, J. (1997) Cancer Res.,
57, 2760-2764.
76.Kyo, S., Takakura, M., Kohama, T., and Inoue, M.
(1997) Cancer Res., 57, 610-614.
77.Saito, T., Schneider, A., Martel, N., Mizumoto,
H., Bulgaymoerschel, M., Kudo, R., and Nakazawa, H. (1997) Biochem.
Biophys. Res. Commun., 231, 610-614.
78.Rhyu, M. S. (1995) J. Natl. Cancer Inst.,
87, 884-894.
79.Sommerfeld, H. J., Meeker, A. K., Posadas, E. M.,
and Coffey, D. S. (1995) Cancer, 75, 2027-2035.
80.Meeker, A. K., Sommerfeld, H. J., and Coffey, D.
S. (1996) Endocrinology, 137, 5743-5746.
81.Counter, C. M., Gupta, J., Harley, C. B., Leber,
B., and Bacchetti, S. (1995) Blood, 85, 2315-2320.
82.Hiyama, K., Hirai, Y., Kyoizumi, S., Akiyama, M.,
Hiyama, E., Piatyszek, M. A., Shay, J. W., Ishioka, S., and Yamakido,
M. (1995) J. Immunol., 155, 3711-3715.
83.Chiu, C. P., Dragowska, W., Kim, N. W., Vaziri,
H., Yui, J., Thomas, T. E., Harley, C. B., and Lansdorp, P. M. (1996)
Stem Cells, 14, 239-248.
84.Igarashi, H., and Sakaguchi, N. (1996)
Biochem. Biophys. Res. Commun., 219, 649-655.
85.Weng, N. P., Levine, B. L., June, C. H., and
Hodes, R. J. (1996) J. Exp. Med., 183, 2471-2479.
86.Bodnar, A. G., Kim, N. W., Effros, R. B., and
Chiu, C. P. (1996) Exp. Cell. Res., 228, 58-64.
87.Buchkovich, K. J., and Greider, C. W. (1996)
Mol. Biol. Cell, 7, 1443-1454.
88.Igarashi, H., and Sakaguchi, N. (1997)
Blood, 89, 1299-1307.
89.Pan, C. G., Xue, B. H., Ellis, T. M., Peace, D.
J., and Diaz, M. O. (1997) Exp. Cell Res., 231,
346-353.
90.Yamada, O., Motoji, T., and Mizoguchi, H. (1996)
Biochim. Biophys. Acta. Mol. Cell Res., 1314,
260-266.
91.Yashima, K., Piatyszek, M. A., Saboorian, H. M.,
Virmani, A. K., Brown, D., Shay, J. W., and Gazdar, A. F. (1997) J.
Clin. Pathol., 50, 110-117.
92.Yasumoto, S., Kunimura, C., Kikuchi, K., Tahara,
H., Ohji, H., Yamamoto, H., Ide, T., and Utakoji, T. (1996)
Oncogene, 13, 433-439.
93.Harle-Bachor, C., and Boukamp, P. (1996) Proc.
Natl. Acad. Sci. USA, 93, 6476-6481.
94.Ramirez, R. D., Wright, W. E., Shay, J. W., and
Taylor, R. S. (1997) J. Invest. Dermatol., 108,
113-117.
95.Hsiao, R., Sharma, H. W., Ramakrishnan, S.,
Keith, E., and Narayanan, R. (1997) Anticancer Res., 17,
827-832.
96.Piatyszek, M., Kim, N., Weinrich, S., Hiyama, K.,
Hiyama, E., Wright, W., and Shay, J. (1995) Meth. Cell Sci.,
17, 1-15.
97.Wright, W. E., Piatyszek, M. A., Rainey, W. E.,
Byrd, W., and Shay, J. W. (1996) Dev. Genet., 18,
173-179.
98.Wright, W. E., Shay, J. W., and Piatyszek, M. A.
(1995) Nucleic Acids Res., 23, 3794-3795.
99.Kim, N., and Wu, F. (1997) Nucleic Acids
Res., 25, 2595-2597.
100.Sallinen, P., Miettinen, H., Sallinen, S. L.,
Haapasalo, H., Helin, H., and Kononen, J. (1997) Am. J. Pathol.,
150, 1159-1164.
101.Krupp, G., Kuhne, K., Tamm, S., Klapper, W.,
Heidorn, K., Rott, A., and Parwaresch, R. (1997) Nucleic Acids
Res., 25, 919-921.
102.Murnane, J. P., Sabatier, L., Marder, B. A.,
and Morgan, W. F. (1994) EMBO J., 13, 4953-4962.
103.Rogan, E. M., Bryan, T. M., Hukku, B., Maclean,
K., Chang, A. C., Moy, E. L., Englezou, A., Warneford, S. G.,
Dalla-Pozza, L., and Reddel, R. R. (1995) Mol. Cell. Biol.,
15, 4745-4753.
104.Bryan, T. M., Englezou, A., Gupta, J.,
Bacchetti, S., and Reddel, R. R. (1995) EMBO J., 14,
4240-4248.
105.Bryan, T. M., Marusic, L., Bacchetti, S.,
Namba, M., and Reddel, R. R. (1997) Hum. Mol. Genet., 6,
921-926.
106.Henderson, S., Allsopp, R., Spector, D., Wang,
S. S., and Harley, C. (1996) J. Cell. Biol., 134,
1-12.
107.Feng, J., Funk, W. D., Wang, S. S., Weinrich,
S. L., Avilion, A. A., Chiu, C. P., Adams, R. R., Chang, E., Allsopp,
R. C., Yu, J., et al. (1995) Science, 269,
1236-1241.
108.Blasco, M. A., Rizen, M., Greider, C. W., and
Hanahan, D. (1996) Nature Genet., 12, 200-204.
109.Avilion, A. A., Piatyszek, M. A., Gupta, J.,
Shay, J. W., Bacchetti, S., and Greider, C. W. (1996) Cancer
Res., 56, 645-650.
110.Nakamura, T. M., Morin, G. B., Chapman, K. B.,
Weinrich, S. L., Andrews, W. H., Linger, J., Harley, C. B., and Cech,
T. R. (1997) Science, 277, 955-958.
111.Meyerson, M., Counter, C. M., Eaton, E. N.,
Ellisen, L. W., Steiner, P., Caddle, S. D., Ziaugra, L., Beijersbergen,
R. L., Davidoff, M. J., Liu. Q., Bacchetti, S., Haber, D. A., and
Weinberg, R. A. (1997) Cell, 90, 785-795.
112.Kanazawa, Y., Ohkawa, K., Ueda, K., Mita, E.,
Takehara, T., Sasaki, Y., Kasahara, A., and Hayashi, N. (1996)
Biochem. Biophys. Res. Commun., 225, 570-576.
113.Norton, J. C., Piatyszek, M. A., Wright, W. E.,
Shay, J. W., and Corey, D. R. (1996) Nature Biotechnol.,
14, 615-619.