REVIEW: What Ultrastable Globular Proteins Teach Us about Protein Stabilization
R. Jaenicke
Institut für Biophysik und Physikalische Biochemie, Universität Regensburg, Universitätsstraße 31, D-93040 Regensburg, Germany; fax: +49 (0) 941-943-2813; E-mail: jaenicke@biologie.uni-regensburg.de
Received June 28, 1997
Proteins, due to their delicate balance of stabilizing and destabilizing interactions, are only marginally stable if physiological conditions are considered as the standard state. Enhanced intrinsic stability of "ultrastable" proteins, e.g., from extremophiles, requires only minute local structural changes. Thus, general strategies of stabilization are not available for temperature, pH, salt, or pressure adaptation. Mechanisms of enhanced thermal stability involve improved packing or docking of structural elements (domains, subunits), as well as specific local interactions, e.g., networks of ion pairs. Relating the structure and stability of eye lens crystallins (which do not undergo any turnover during the life time of an organism), point mutations, nicking and swapping of domains, grafting of linker peptides between domains, and denaturation-renaturation allowed the cumulative nature of protein stability and its relation to the hierarchy of protein structure and folding to be established. In this review, recent results for crystallins and enzymes from hyperthermophiles will be discussed as models to illustrate mechanisms of protein stabilization.
KEY WORDS: association, chaperons, crystallins, domains, extremophiles, eye lens proteins, folding, fragments, glycolytic enzymes, linker peptide, oligomeric proteins, self-assembly, stabilization, thermophilia
Proteins are multifunctional in the sense that their specific amino acid sequence simultaneously determine: a) the self-organization of the linear polypeptide chain to form a unique three-dimensional structure; b) a specific function and its regulation, and c) degradation. Correspondingly, evolution has to compromise between rigidity (i.e., stability) on one hand, and flexibility (i.e., function, regulation, and turnover) on the other. The way nature reaches this goal is that the Gibbs free energy of stabilization of proteins is of the order of 50 kJ/mole, i.e., the equivalent of only a few weak interactions. This holds in spite of the fact that the estimated total energy of the attractive and repulsive interatomic interactions is of the order of 107 kJ/mole [1, 2].
As Anson and Anfinsen have pointed out, the "translation" of the one-dimensional information, encoded in the amino acid sequence, into the spatial configuration occurs autonomously. In the course of the spontaneous reaction, the folding polypeptide chain gains stability at different levels of Linderström--Lang hierarchy of protein structure. From the mechanistic point of view, protein self-organization does not proceed strictly sequentially, from secondary structural elements to the native tertiary and quaternary structure. The reason is that the tertiary contacts themselves select the correct local structures and stabilize them, such that local structures with low stability and high folding and unfolding rates are in rapid equilibrium early in folding. What seems clearly established is the observation that structure formation starts from next-neighbor interactions which, in the time range of milliseconds, generate stretches of secondary structure. Subsequently, non-local interactions lead to metastable supersecondary structures and subdomains; these (still within fractions of a second), collapse into the native-like "molten globule state" of domains as independent folding units or, at least, separable structural entities. The corresponding shuffling processes seem to occur along a more or less "ragged energy landscape" by a limited number of pathways, with late rate-limiting steps. If, at this level, there is still significant hydrophobic surface area exposed to the aqueous solvent, folding and association will end up in the geometrically and stoichiometrically unique quaternary structure. Domain pairing and subunit recognition are highly specific, even in the presence of excess heterologous proteins. Both contribute significantly to the intrinsic stability of proteins [3, 4].
In considering folding as the primary function of the nascent (or refolding) polypeptide chain, one has to keep in mind that interactions with other proteins such as chaperons and folding catalysts, or membrane components may serve to gear the process in terms of specific recognition, morphopoiesis, addressing, chemical modification, etc., finally providing the native state. This serves its specific function until the end of the "protein life cycle" when spontaneous unfolding or ubiquitinylation, and subsequent proteolytic degradation refill the cellular amino acid pool for either metabolism or renewed translation. In quantitative terms, the life span of proteins reaches from minutes (e.g., insulin) to the life span of organisms (e.g., eye lens crystallins).
The survey of the fundamentals of stability and folding needs to be discussed in the context of the enormous adaptive capacity of life on earth: except for centers of volcanic action, life on earth is ubiquitous. The biosphere (including zones where cryptobiosis prevails) refers to the whole surface of the globe, the soil to a depth of 10-20 m, the oceans down to the abyssal regions, the atmosphere and the stratosphere [2]. Translated into the language of folding and function, this means that cells and their protein inventory have to cope with extremes of physical conditions close to the dissociation energies of strong covalent chemical bonds. Regarding the long-term stability, similar challenges are met (e.g., the life-long stability of certain eye lens proteins [5]). Evidently, nature provides us with biotopes and organelles that stand out for extreme adaptive pressure. Their protein repertoire requires high stability, either intrinsic or extrinsic.
EXTREMOPHILES
In addressing the question how extremophiles adjust to extremes of physical conditions, it is important to note that, apart from mutational adaptation at the DNA and protein level, extrinsic factors such as prosthetic groups may assist in the stabilization of proteins. In addition, they are known to stabilize both the native state and intermediates on the pathway of folding and association [4, 6, 7]. The same holds true for the conjugation of proteins, e.g., with carbohydrates or nucleic acids. Using internal, core-glycosylated, and external invertase from yeast (with 0, 34, and 68% carbohydrate content) as a model, three important observations have been made: 1) glycosylation causes significant stabilization and solubilization of proteins; 2) it does not affect the mechanism of folding and association, and 3) due to its potent solubilization capacity, the carbohydrate part protects the attached protein moiety from (thermal) aggregation, exhibiting a chaperon-like effect in the kinetic partitioning between folding and aggregation [8, 9]. In the case of nucleoproteins, protein--protein and protein--nucleic acid interactions lead to enhanced stability; the assignment of defined stability increments to the constituent parts is ambiguous.
Under various stress conditions, including chill acclimation, drought and salinity, the protective high local concentration of sugar moieties observed in glycoproteins is mimicked by the up-regulation of the synthesis of polyhydroxy compounds or metabolites. Corresponding stress responses have been observed in bacteria, plants and even small animals [10-12]. For thermophiles, their stabilizing effect on proteins is a consequence of preferential solvation, whereas in yeast or halophiles polyols serve as osmolytes [13-15]; in drought-tolerant desiccation plants, lysine-, serine- and threonine-rich "resurrection proteins" seem to serve as mimics for intracellular water under conditions of severe water loss [16].
The previous examples illustrate the fundamentally different ways how organisms can cope with extremes of physical parameters: they may avoid the stress, e.g., by neutralizing anomalous pH values or balancing high salt concentrations through isosmotic concentrations of compatible solutes, or they may tolerate it or adapt to it by mutative changes, so that all the cell’s constituents are resistant against the specific variable. Because of the isothermal and isobaric conditions in a given habitat, evidently the entire cell inventory of thermophiles and barophiles has to be adapted, to the extent that both require the extreme condition for their life cycle. In the case of hyperthermophiles, the range of optimal growth is between 80 and 110°C. The respective upper limits of the various parameters in the biosphere are: for temperature, ~120°C; for hydrostatic pressure, 120 MPa (1200 bar); for water activities at high salt concentrations, .0.6; for [H+], pH values between 0 and 11 with the cytosolic pH regulated close to neutrality [2].
Limits of Thermal Stability
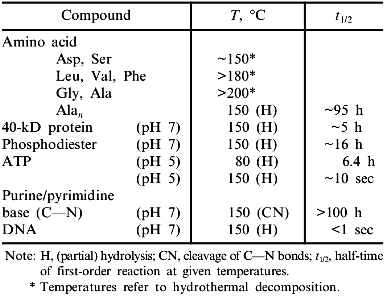
Considering the temperature limit of growth in the biosphere, two observations have to be kept in mind: 1) proteins from extremophiles consist exclusively of the canonical natural amino acids; 2) at temperatures beyond 110°C, proteins and their constituent amino acids undergo chemical modification. Thus, hyperthermophiles must either compensate for protein degradation by enhanced synthesis, or they must have evolved repair mechanisms or extrinsic protection strategies, e.g., using compatible solutes. Oxidative degradation affects mainly cysteine and methionine, whereas asparagine and glutamine undergo deamidation. In the case of glycoproteins, the Maillard reaction may serve as a paradigm to illustrate the complexity of the chemistry involved [7, 17, 18]. Little is known about the detailed mechanisms involved in the hydrothermal degradation of amino acids, even less about repair and protection. Ions, cofactors, metabolites, or conjugation with non-proteinaceous components have been shown to contribute to protein stability, to the extent that occasionally the denaturation temperature of thermally unstable proteins may be shifted to the upper temperature limit of hydrothermal degradation [10, 11, 19]. Apart from covalent modifications, two more limiting factors need consideration: 1) the increase in thermal energy interferes with the weak interactions responsible for protein folding and association; this holds especially for the effect of hydrophobic hydration which is known to vanish at temperatures beyond 120°C (see [2]); 2) (non-catalyzed) hydrolysis of ATP in aqueous solution occurs at temperatures below 140°C [20], setting a limit to anabolic reactions, especially to the resynthesis of biomolecules (Table 1).
Table 1. Upper limit of thermal stability
of biomolecules in aqueous solutions
Forces
The stability of proteins is the cumulative effect of local interactions between side chains, the polypeptide backbone, domains, and subunits. At optimal physiological temperatures of (hyper)thermophiles, contributions to the stability of proteins originate mainly from hydrophobic and coulomb interactions. Both become stronger with increasing temperature, because of their entropic origin, on one hand, and the temperature dependence of the dielectric constant, on the other. Apart from this classical explanation, advances in the measurement and theory of hydration have shown that temperature-induced recognition of polar rather than hydrophobic residues may be involved in macromolecular assembly owing to complementary patterns of surface polar groups. As shown by direct measurements of temperature dependent intermolecular forces, the energies involved can be high (~0.6 kcal/mole per polar group) [21]. In defining surface polar groups, ionic networks seem to contribute significantly to protein stability (see below). Their distinction from clusters of hydrogen bonds, e.g., comparing
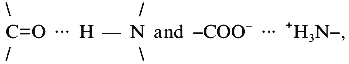
LEVELS OF STABILIZATION
In contrast to the unpredictable local contributions to protein stability, protein fragments, as well as domains and subunits, are often correlated with well-defined structural increments of DeltaGstab. With respect to the stability of supersecondary structures or subdomains, proteins have been shown to be cooperative systems showing mutual stabilization of structural elements and additivity of increments of stabilization. Using thermolysin (316 amino acids, all-helical monomer) to determine the length of a polypeptide chain which still forms native-like supersecondary structure, the N-terminal 62-residues three-helix bundle still exhibits much of its native secondary structure. At a size of 20 residues, intermolecular interactions dominate, causing aggregation instead of proper fragment recognition or native-like structure formation [22].
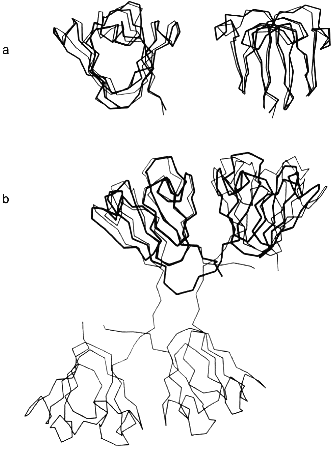
At the next-higher level of the structural hierarchy of proteins, domains show intrinsic stability as independent structural entities; apart from "folding by parts", they often gain stability from domain interactions [2, 4]. The eye lens protein gammaB-crystallin may be taken as an example. The protein consists of two topologically identical 10 kD domains, exclusively beta-sheet (Fig. 1a). As mentioned, it does not undergo significant degradation during our lifetime. In the case of gammaB-crystallin, the N-terminal domain shares the extreme stability of the complete molecule, whereas the C-terminal domain is less stable, giving rise to a bimodal equilibrium transition in the presence of urea as a denaturant [23, 24]. The underlying three-state mechanism N <--> I <--> U (with N and U as native and denatured states, and I as intermediate with one domain unfolded and the other still native) is confirmed by equilibrium denaturation transitions, as well as kinetic folding/unfolding experiments with the natural two-domain protein and its isolated domains (Table 2).
Table 2. Urea-induced unfolding transitions of wild-type and mutant gammaB-crystallins monitored by fluorescence emission at pH 2.0, 20°CFig. 1.Structure of betagamma-crystallins from the eye lens. a) The all-beta C- and N-terminal domains of bovine gammaB-crystallin exhibit high structural similarity: the two 90° views show the superposition of the Calpha-backbones (N-terminal domain, thin line; C-terminal domain, thick line). b) Calpha-backbones of the polypeptide chains of gammaB-crystallin (thick line) and betaB2-crystallin (thin line) illustrating "domain swapping" [3].

In analyzing the equilibrium transitions, the second phase is found to coincide with the folding/unfolding of the isolated N-terminal fragment, proving this half of the entire molecule to exhibit an excess stability of DeltaDeltaGN-->U ~ 15 kJ/mole; the lower stability of the C-terminal half shows that, under the given conditions, domain interactions contribute significantly to the overall stability of the protein. At first glance, it is surprising that the two topologically closely related domains show such drastic deviations in their stability properties. However, considering the difference in net charge between the two domains at pH 2, it is clear that the electrostatic potential adds to the destabilization of the C-terminal half of the molecule in the presence of the chaotropic agent [25].
The previously mentioned contributions of domain interactions to the anomalous overall stability of gammaB-crystallin has been confirmed by a variety of experiments (see Table 2).
1. Based on denaturation data with urea as denaturant, the stabilities of the domains in complete gammaB-crystallin and those of the isolated recombinant domains differ significantly, pointing to a mutual stabilization of the order of 10-20 kJ/mole [26].
2. There is a hydrophobic cluster in the interface between the two domains in which Met-43, Phe-56, Ile-81 (N-terminal), and Val-132, Leu-145, Val-170 (C-terminal) are in van der Waals contact. Substituting Ala, Trp, or Asp for Phe-56 affects the stability, as one would predict from both the hydrophobicity and packing of the interdomain space: creating a crevice, or putting an ionizable residue into the non-polar interface (as in the case of Ala or Asp), destabilizes the protein; replacing Phe by Trp is less effective [27]. Surprisingly, the two interface arginine residues 79 and 147 which face each other from a distance of 3.3 Å, do not cause destabilization by charge repulsion; rather they contribute to the stability of the two-domain structure (see mutant R79E, Table 2).
3. gammaB- and betaB2-crystallins are distinct mainly by their linker peptides connecting the two domains in each polypeptide chain. As illustrated in Fig. 1b, "swapping" of the domains allows switching from interdomain to intersubunit interactions [3, 26]. As a consequence, gammaB-crystallin, with its V-shaped connecting peptide between the domains, is monomeric, whereas betaB2-crystallin, with the linker in an extended conformation, is a homodimer. Assuming the connecting peptide to determine the mode of domain association, one would predict that grafting the extended betaB2 linker into gammaB would lead to a gammaB dimer. However, when hybrids were engineered so that gammaB contained the betaB2 linker and vice versa, monomers were obtained. Thus, in both constructs, the connecting peptide must be twisted to a turn similar to the one in natural gammaB-crystallin [26]. The observation that the betaB2 construct is shifted to the monomer clearly indicates that both the domain interface and the connecting peptide must be involved in the partitioning between intra- and interchain interactions. Which of the two is more important depends, first, on the specific linker configuration, second, on the packing properties of a given tertiary or quaternary structure and, third, on the "local concentration" required for the recognition and mutual stabilization of the domains. The latter condition explains why mixing the N- and C-terminal domains at a 1:1 ratio does not yield "nicked gammaB". In summarizing the results of the above mutant studies, it becomes clear that the tuning of the interactions responsible for the local concentrations of the domains and their recognition is extremely sensitive to marginal alterations of the connecting peptides, on the one hand, and single residues in the domain surface, on the other. Evidently, in the eye lens, the multitude of homologous components with slight structural variations allow the unperturbed accumulation of high protein levels without phase separation or aggregation. In this context it is worth mentioning that molecular crowding contributes to protein stability [28]; in addition, it has been shown that alpha-crystallin, the main protein component of the vertebrate eye lens, is a heat shock protein and serves as a chaperon, keeping misfolding and aggregation at a minimum [29].
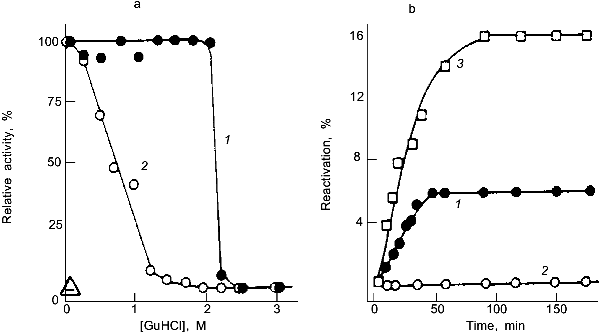
Quaternary interactions of subunits in oligomeric or multimeric proteins are the equivalent of the mutual stabilization of domains in multidomain proteins. Their stabilizing effect may be illustrated taking lactate dehydrogenase (LDH) as an example. Using guanidine denaturation, the intrinsic stability of the enzyme has been shown to increase steadily in proceeding from domain fragments to domains and domain-assemblies, and ending with monomers, dimers, and tetramers. The latter, as a dimer of dimers, is highly stable, the "proteolytic dimer" (lacking the N-terminal 10 amino acids) requires structure-making salt to exhibit activity, the monomer shows native-like structure only as a short-lived folding intermediate on the pathway of reconstitution. The separate domains are unstable, but still sufficiently well-defined in their conformation to recognize one another and to pair correctly; in joint reconstitution experiments, they lead to partial renaturation, exhibiting a mutual "chaperon effect" (Fig. 2) [30].
There are cases in which stabilization due to quaternary structure formation goes to the extreme, e.g., viruses, chromatin, ferritin, bacterial surface layers. In these examples, the monomers or separate constituents have average size, stability and flexibility, and perform their morphopoietic or catalytic functions without being inhibited with respect to translocation, targeting, processing, etc. It is the assembly that is responsible for their anomalous stability. Occasionally, proteins from hyperthermophiles have been found to exhibit higher levels of quaternary structure compared to their mesophilic counterparts, suggesting a correlation between subunit association and thermophilic adaptation [31-33] (see below). As for all other "adaptive strategies", there are examples contradicting this mechanism [7, 34].Fig. 2. Stability and reconstitution of porcine muscle lactate dehydrogenase and its domain fragments. a) Deactivation of the native tetramer (1) and the "proteolytic dimer" lacking the N-terminal decapeptide (2); as indicated by the reactivation kinetics, the structured monomer is unstable and inactive (Delta) [4]. b) Reactivation of the "proteolytic dimer" (3) and its separate 14- and 21-kD domain fragments, F14 and F21, after denaturation in 6 M guanidine hydrochloride; 1) joint reconstitution of F14 and F21; 2) separate reconstitution and subsequent mixing of F14 and F21 [30].
THERMAL STABILITY OF PROTEINS FROM HYPERTHERMOPHILES
The central issue in the adaptation of enzymes to extremes of physical conditions is the conservation of their functional state, which is characterized by a well-balanced compromise of stability and flexibility. Considering the marginal extra free energy, DeltaDeltaGN-->U, characteristic for ultrastable proteins, it is apparent that enhanced stability may be accomplished by a vast number of mechanisms. Well-established observations are as follows.
1. Proteins from thermophiles commonly exhibit high intrinsic stability; as in the case of proteins from mesophiles, this becomes marginal at their optimum temperature.
2. The increased rigidity of thermophilic proteins becomes evident from a wide variety of observations, e.g., decreased H/D exchange rates of amide protons, enhanced activity at low denaturant concentrations, and increased resistance against proteolysis and denaturants.
3. Available structural data prove that proteins from thermophiles are closely similar to their mesophilic counterparts regarding their basic topologies and enzyme mechanisms.
4. The correlation of sequence alterations with changes in thermal stability is complex and does not allow for general rules [35].
5. High-resolution X-ray and NMR data confirm the cumulative mechanism of protein stability, in individual cases allowing certain principles of thermal stabilization to be extracted.
The following examples refer to a small number of proteins from the hyperthermophilic bacterium Thermotoga maritima selected to illustrate either the "individual character" of proteins in general or possible specific mechanisms of protein stabilization. Thermotogales are among the earliest (and most thermophilic) representatives in the phylogeny of the bacterial domain of life [7, 36]. Their characteristic outer envelope, the "toga", consists of a porin-like trimeric protein that not only protects the murein cell wall but, at the same time, serves as a scaffold integrating hydrolases and certain transporters ([37, 38], H. Schurig, D. Wassenberg, and W. Liebl, personal communication).
Stress Proteins and Protein Folding
Surprisingly, hyperthermophiles such as Thermotoga maritima contain cold shock and heat shock proteins. The first exhibit structural similarity with all known homologs, but their function is still enigmatic; the latter resemble other members of the Hsp60 family and show chaperon activity [39, 40]. What is relevant in the present context is that folding assistance (at least in vitro) becomes significant only at temperatures close to the optimal growth temperature of the bacterium, supporting the idea that mesophilic and thermophilic proteins under their respective physiological conditions are in corresponding states [2]. Little is known about the detailed structure and the mechanism. That in vivo the protein actually serves as a molecular chaperon is suggested by its expression under thermal stress, and its structural homology with E. coli GroEL including its ATPase activity [40]. Using in vitro reconstitution experiments to simulate the in vivo reaction, the chaperon is found to improve the yield by shifting the kinetic competition between folding and aggregation toward the native state. Whether seclusion of the folding polypeptide in the interior of the chaperon, or excluded volume effects in the crowded cytosol serve to keep aggregation-competent intermediates below a critical concentration is still an open question [3].
Considering folding per se, the question arises how early intermediates on the folding pathway can cope with temperatures close to the boiling point of water without being trapped by heat aggregation. Early statistical analyses seemed to indicate that thermal adaptation is reflected by an overall increase in hydrophobicity so that thermal stabilization of the collapsed state as the earliest stable intermediate seemed to be the general solution to the problem. Since recent data (including the sequences of the complete genomes of hyperthermophiles) do not confirm the statistics [35], other properties must be important.
Investigating thermal effects on the (re)folding of ultrastable proteins, both in vivo and in vitro, the influence of temperature on protein folding is found to be less pronounced than originally expected.
1. The expression of active proteins from hyperthermophiles in mesophilic hosts can be easily performed 60°C below their physiological temperature [41].
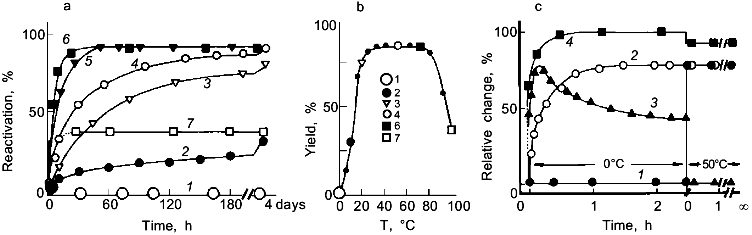
2. As shown in Fig. 3, reconstitution of ultrastable proteins may be accomplished over a wide temperature range. For glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from Thermotoga, even at 100°C, significant reactivation has been observed proving that the thermal stability of the enzyme refers not only to the native state but also to all intermediates in their various states of association along the pathway of folding and oligomerization [42].
3. There is no indication for cold denaturation at 0°C (more than 80°C below the in vivo temperature). On the other hand, there is no reactivation at 0°C. Instead, the enzyme is trapped as an inactive "cold intermediate", Ic, which can be fully activated to the native state by shifting the temperature to 50°C (Fig. 3c). Ic is a tetramer with native-like secondary structure; its aromatic residues are still exposed to the aqueous solvent. Thus, even 80-90°C below the in vivo temperature, Thermotoga GAPDH folds to a native-like structure [43].
Glycolytic Enzymes from Thermotoga maritimaFig. 3. Temperature effect on the reactivation of GAPDH from Thermotoga maritima after preceding denaturation in 6 M guanidine hydrochloride. a) Reactivation kinetics after dilution in 50 mM phosphate buffer, pH 7, at 0 (1), 10 (2), 20 (3), 30 (4), 50 (4), 70 (6), and 100°C (7). b) Temperature dependence of the yield of reactivation after 4 days. c) Formation of the native-like cold intermediate, Ic, upon renaturation at 0°C; shifting the temperature to 50°C after 2.5 h recovers the native state. Reactivation at 0 (1) and 50°C (2); 3) regain of native fluorescence emission (F320); 4) far-UV circular dichroism ([theta]222) [42, 43].
Thermotoga maritima gains metabolic energy by substrate-level phosphorylation rather than respiration so that most glycolytic enzymes are expressed at levels sufficiently high for purification and physical characterization. Detailed studies on triosephosphate isomerase (TIM), GAPDH, phosphoglycerate kinase (PGK), enolase, and LDH have been reported in a recent review [7]. Significant differences between the hyperthermophilic enzymes and their mesophilic counterparts refer to the packing density, a reduced surface/volume ratio, increased intersubunit hydrophobicity, anomalous states of association, fusion of otherwise separate genes, etc. Limited space allows only two examples to be discussed.
1. TIM, PGK, and PGK--TIM fusion protein. Thermotoga does not possess a separate gene for TIM. Instead, TIM activity is observed in a fusion protein coding for PGK--TIM [44]. Within the bifunctional polypeptide chain, at its C-terminal end, TIM occupies a length of 27 kD, which corresponds to the subunit size known for the enzyme from other sources. The three-dimensional structure is the common (alphabeta)8 barrel, as predicted from sequence comparison and homology modeling (R. K. Wierenga, personal communication).
PGK shows anomalous properties: apart from the bifunctional 280 kD tetramer with PGK and TIM activity, it occurs as the common-size 45 kD monomer; a frame-shift upstream of its stop codon is responsible for the linkage of the two genes. The fusion gene has been cloned in E. coli to the effect that in the host the same frame-shift occurs as in Thermotoga. Whether the underlying mechanism is related to a transcriptional or translational event needs still to be worked out.
The spatial structure of monomeric PGK is closely similar to that described for mesophilic and moderately thermophilic homologs. Binding the substrates (3-phosphoglycerate and adenylylimidodiphosphate (AMP-PNP)) leads to a closed configuration of the enzyme proving a domain rotation by ~15° to be involved in the enzyme mechanism [45, 46].
The analysis of the three-dimensional structure of the PGK--TIM fusion protein has not been completed yet so that, at present, no unambiguous information is available with respect to the relative orientation of the active sites of TIM and PGK in the tetrameric complex. Indirect evidence favors the idea that the TIM dimer forms the core with PGK as peripheral modules (G. Auerbach and R. K. Wierenga, personal communication).
Thermal analysis and chemical denaturation of the PGK--TIM fusion protein and its recombinant separate enzymes, as well as PGK domains, indicate that domain interactions and subunit interactions contribute to DeltaGN-->U of the PGK--TIM fusion protein ([47], K. Zaiss, personal communication). Surprisingly, the recombinant or separate domains of PGK exhibit increased stabilities compared to their contributions to the free energy of stabilization for the complete monomer [48, 49].
The denaturation of the fusion protein shows bimodal characteristics indicating independent folding of its constituents parts. The reaction is only partially reversible, probably due to wrong pairing of domains and subunits. As taken from the unaltered thermal transition of monomeric PGK and the enzyme integrated into the fusion protein, the change in quaternary structure has no effect on the intrinsic stability. This is different for TIM: comparing the properties of the TIM portion of the PGK--TIM fusion protein with those of the recombinant isolated TIM dimer, both the intrinsic stability and the catalytic efficiency of the enzyme are found to be enhanced in the tetrameric fusion protein. Since there is no common intermediate in the mechanism of the two coupled enzymes, substrate channelling cannot be the reason for the improvements; what might be the evolutionary advantage of the gene fusion is still enigmatic [47].
2. GAPDH. GAPDH has been a paradigm of protein stabilization since J. I. Harris’ pioneering studies on the Bacillus stearothermophilus enzyme and its comparison with homologous GAPDHs from other organisms [50]. The Thermotoga enzyme expands the temperature scale beyond the boiling point of water. The physicochemical and enzymic properties of the enzyme have recently been reviewed [51]. The following general findings are of importance.
1. In spite of the high sequence identity of the enzymes from various bacteria (~60% with only 8% non-conservative exchanges), searching for essential substitutions is not practicable.
2. The X-ray structure yields a topology closely similar to the enzyme from lobster and Bacillus stearothermophilus [52]; differences between the hyperthermophilic protein and its less stable counterparts are quantified by mean positional errors of the order of 0.3 Å, and r.m.s. deviations of superimposed Calpha positions in different domains of <1 Å, so that no general conclusions with respect to thermal stability and stabilization can be given. Insertions and deletions are found exclusively in loops in the surface of the molecule. Here, the most significant difference is a network of charged residues involved in peripheral ion pairs. Additional stability increments come from improved hydrophobic interactions, especially in the subunit interfaces.
3. Under standard conditions, Thermotoga GAPDH exhibits anomalous rigidity due to improved packing of the polypeptide chain; it vanishes at elevated temperature, i.e., under "corresponding state" conditions [2, 51].
Temperature-Sensitive Mutants
In order to identify specific amino acid residues responsible for the high intrinsic stability of ultrastable proteins, either selection for mutants with differing stabilities or site-directed mutagenesis may be applied. In the case of Thermotoga GAPDH, both approaches have flaws: on one hand, selection for mutants with enhanced stability will soon reach the limit of growth due to the hydrothermal degradation of amino acids; on the other hand, there is an impracticably large number of exchanges and combinations of exchanges, given the above sequence identity of ~60%. Therefore, attempts were made to go in the inverse direction, i.e., to either generate or select for temperature-sensitive mutants [53]. The second alternative was unsuccessful because point mutations caused enhanced expression rather than decreased stability of the natural enzyme.
Site-directed mutagenesis focused on ion pairs or ion pair networks that are present in Thermotoga GAPDH but not in the Bacillus stearothermophilus enzyme. Their removal might shift the limit of stability to lower temperatures. Three types of ion pairs were chosen: 1) non-conserved charges in Thermotoga GAPDH that are not present in the Bacillus stearothermophilus enzyme; 2) ion pairs in the S-loop of the substrate binding domain involved in tertiary and quaternary contacts in the enzyme; 3) highly conserved salt bridges involved in arginine charge clusters in the NAD+-binding domain. All three types of mutation sites are located in the periphery of the tetrameric enzyme. Mutations of the first category were found to have no effect. In the second case, loop mutations caused a decrease in stability, attributable to the uncoupling of the domains, but not to changes in the tetramer --> monomer transition, even at low temperatures and concentrations. In the third case, the Arg-10 charge cluster involves at the same time one intersubunit and two intrachain ion pairs; in addition, the backbone NH-group forms one out of eight H-bonds responsible for NAD+ binding. Replacing Arg-10 by Met or Lys (to keep the backbone in register and to explore how critical the charge distances are) led to the uncoupling of guanidine hydrochloride deactivation (due to NAD+ release) and denaturation, shifting the latter to lower guanidine concentrations. In the case of the Arg-20-->Ala and Arg-20-->Asn mutations, the kinetic stability of the enzyme is affected: the temperature of half-denaturation (after 1 h) is shifted from 96 to 89°C. Thus, the ionic network around Arg-20 significantly raises the activation barrier against thermal unfolding, causing an increase of the half-life of the enzyme at the physiological temperature of the hyperthermophile.
Taken together, changes in peripheral ion pairs are not the general strategy of thermal stabilization. Even after carefully selecting specific loci for site-directed mutagenesis on the basis of multiple sequence alignment and homology modeling, the results are unpredictable. Each mutant exhibits new and unexpected properties. Recalling that the overall stability of proteins is a marginal difference between large contributions of attractive and repulsive forces plus the corresponding entropy contributions, this result is after all not surprising: proteins are individuals.
Work performed in the author’s laboratory was generously supported by the Deutsche Forschungsgemeinschaft, the Fonds der Chemischen Industrie, the Alexander von Humboldt Stiftung, and the Fogarty International Center for Advanced Studies. Fruitful discussions with Drs. N. Beaucamp, R. Glockshuber, E.-M. Mayr, S. Palme, R. Rudolph, H. Schurig, R. Seckler, C. Slingsby, and K. O. Stetter are gratefully acknowledged.
REFERENCES
1.Baldwin, R. L. and Eisenberg, D. (1987) in
Protein Engineering (Oxender, D. E., and Fox, C. F., eds.) Liss,
New York, pp. 127-148.
2.Jaenicke, R. (1991) Eur. J. Biochem.,
202, 715-728.
3.Jaenicke, R. (1996) Curr. Top. Cell. Regul.,
34, 209-314.
4.Jaenicke, R. (1987) Progr. Biophys. Mol.
Biol., 49, 117-237.
5.Jaenicke, R. (1996) FASEB J., 10,
84-92.
6.Risse, B., Stempfer, G., Rudolph, R., and Jaenicke,
R. (1992) Protein Sci., 1, 1699-1718.
7.Jaenicke, R., Beaucamp, N., Schurig, H., and
Ostendorp, R. (1996) Adv. Protein Chem., 48,
181-269.
8.Kern, G., Schülke, N., Schmid, F. X., and
Jaenicke, R. (1992) Protein Sci., 1, 120-131.
9.Kern, G., Kern, D., Jaenicke, R., and Seckler, R.
(1993) Protein Sci., 2, 1862-1868.
10.Hensel, R., and König, H. (1988)
FEMS-Microbiol. Lett., 49, 75-79.
11.Huber, R., Kurr, M., Jannasch, H.-W., and
Stetter, K. O. (1989) Nature (London), 342, 833-834.
12.Franks, F. (1990) Phil. Trans. Roy. Soc.
Lnd., B326, 517-533.
13.Timasheff, S. N. (1993) Annu. Rev. Biophys.
Biomol. Struct., 22, 67-97.
14.Blomberg, A., and Adler, L. (1992) Adv.
Microbiol. Physiol., 33, 145-212.
15.Lisse, T., Bartels, D., Kalbitzer, H. R., and
Jaenicke, R. (1996) Biol. Chem., 377, 555-561.
16.Franks, F., and Grigera, J. R. (1990) in Water
Science Reviews (Franks, F., ed.) Vol. 5, Cambridge University
Press, Cambridge, New York, Sidney, pp. 187-289.
17.Bernhardt, G., Lüdemann, H.-D., Jaenicke,
R., König, H., and Stetter, K. O. (1984) Naturwissen.,
71, 583-586.
18.Barrett, G. C. (1985) in Chemistry and
Biochemistry of Amino Acids (Barrett, G. C., ed.) Chapman and Hall,
London, New York, pp. 354-375.
19.Scholz, S., Sonnenbichler, J., Schäfer, W.,
and Hensel, R. (1992) FEBS Lett., 306, 239-242.
20.Leibrock, E., Bayer, P., and Lüdemann, H.-D.
(1995) Biophys. Chem., 54, 175-180.
21.Leikin, S., and Parsegian, V. A. (1994)
Proteins: Struct. Funct. Genet., 19, 73-76.
22.Vita, C., Jaenicke, R., and Fontana, A. (1989)
Eur. J. Biochem., 183, 513-518.
23.Jaenicke, R. (1994) Naturwissen.,
81, 423-429.
24.Rudolph, R., Neßlauer, R., Siebendritt, R.,
Sharma, A. K., and Jaenicke, R. (1990) Proc. Natl. Acad. Sci.
USA, 87, 4625-4629.
25.Mayr, E.-M., Jaenicke, R., and Glockshuber, R.
(1997) J. Mol. Biol., 269, 260-269.
26.Mayr, E.-M., Glockshuber, R., and Jaenicke, R.
(1994) J. Mol. Biol., 35, 84-88.
27.Palme, S., Slingsby, C., and Jaenicke, R. (1997)
Protein Sci., 6, 1529-1536.
28.Zimmerman, S. B., and Minton, A. P. (1993)
Annu. Rev. Biophys. Biomol. Struct., 22, 27-65.
29.Brady, J. P., Garland, D., Duglas-Tabor, Y.,
Robison, W. G., Jr., Groome, A., and Wawrousek, E. F. (1997) Proc.
Natl. Acad. Sci. USA, 94, 884-889.
30.Opitz, U., Rudolph, R., Jaenicke, R., Ericsson,
L., and Neurath, H. (1987) Biochemistry, 26,
1399-1406.
31.Hess, D., Krüger, K., Knappik, A., Halm, P.,
and Hensel, R. (1995) Eur. J. Biochem., 233,
227-237.
32.Kohlhoff, M., Dahm, A., and Hensel, R. (1996)
FEBS Lett., 383, 245-250.
33.Hennig, M., Sterner, R., Kirschner, K., and
Jansonius, J. N. (1997) Biochemistry, 36, 6009-6016.
34.Schurig, H., Rutkat, K., Rachel, R., and
Jaenicke, R. (1995) Protein Sci., 4, 228-236.
35.Böhm, G., and Jaenicke, R. (1994) Int. J.
Pept. Protein Res., 43, 97-106.
36.Stetter, K. O. (1996) FEMS Microbiol.
Rev., 18, 149-158.
37.Rachel, R., Wildhaber, I., Stetter, K. O., and
Baumeister, W. (1988) in Crystalline Bacterial Surface Layers
(Sleytr, U. B., Messner, P., Pum, D., and Sára, M., eds.)
Springer Verlag, Berlin, pp. 83-86.
38.Schumann, J., Wrba, A., Jaenicke, R., and
Stetter, K. O. (1991) FEBS Lett., 282, 122-126.
39.Phipps, B. M., Hoffmann, A., Stetter, K. O., and
Baumeister, W. (1991) EMBO J., 10, 1711-1722.
40.Ostendorp, R. (1996) Struktur, Stabilität
und Funktion Extrem Thermophiler Proteine: LDH aus Thermotoga
maritima, Ph. D. Thesis, University of Regensburg.
41.Tomschy, A., Glockshuber, R., and Jaenicke, R.
(1993) Eur. J. Biochem., 214, 43-50.
42.Rehaber, V., and Jaenicke, R. (1992) J. Biol.
Chem., 267, 10999-11006.
43.Rehaber, V., and Jaenicke, R. (1993) FEBS.
Lett., 317, 163-166.
44.Schurig, H., Beaucamp, N., Ostendorp, R.,
Jaenicke, R., Adler, E., and Knowles, J. (1995) EMBO J.,
14, 442-451.
45.Auerbach, G., Jacob, U., Grättinger, M.,
Zaiss, K., Schurig, H., and Jaenicke, R. (1997) Biol. Chem.,
378,327-329.
46.Auerbach, G., Huber, R., Grättinger, M.,
Zaiss, K., Schurig, H., Jaenicke, R., and Jacob, U. (1997)
Structure, 5, 1475-1483.
47.Schurig, H. (1995) Struktur und Funktion von
Thermostabilen Proteinen: TIM, PGK und Enolase aus Thermotoga
maritima, Ph. D. Thesis, University of Regensburg.
48.Beaucamp, N., Schurig, H., and Jaenicke, R.
(1997) Biol. Chem., 378, 679-685.
49.Beaucamp, N., Hofmann, A., Kellerer, B., and
Jaenicke, R. (1997) Protein Sci., 6, 2159-2165.
50.Harris, J. I., and Walker, J. E. (1977) in
Pyridine, Nucleotide-Dependent Dehydrogenases (Sund, H.,
ed.) De Gruyter, Berlin, New York, pp. 43-61.
51.Jaenicke, R. (1996) FEMS Microbiol. Rev.,
18, 215-224.
52.Korndörfer, I., Steipe, B., Huber, R.,
Tomschy, A., and Jaenicke, R. (1995) J. Mol. Biol.,
246, 512-521.
53.Tomschy, A., Böhm, G., and Jaenicke, R.
(1994) Protein Eng., 7, 1471-1478.