REVIEW: Protein Folding Assisted by the GroEL/GroES Chaperonin System
J. Martin
Department of Molecular Biology, Cell Biology and Biochemistry, Brown University, Box G-J2, Providence, RI 02912, USA; fax: (401) 863-1201; E-mail: Jorg_Martin@Brown.edu
 J. Martin |
Received July 3, 1997
The chaperonin system GroEL/GroES assists in the folding of proteins in the bacterial cytosol. Recent applications of biophysical techniques for the structural analysis of GroEL, GroES, and chaperonin-bound protein folding intermediates have provided the basis for understanding the molecular mechanism of GroEL/GroES action. GroEL, a double-ring complex, binds unfolded proteins at its inner ring surface. Protein folding proceeds in the central cavity of GroEL, after dissociation of the polypeptide has been triggered by ATP hydrolysis in GroEL. Premature release of unfolded protein into external solution is prevented by binding of the cofactor GroES on top of the GroEL cylinder, resulting in an enclosed cage. Upon ATP-dependent dissociation of GroES, substrate protein is eventually released from GroEL in a native or native-like conformation. While current in vitro results about the structure, function, and molecular mechanism of GroEL/GroES-assisted protein folding have led to a quite detailed picture of this complex process, the extent to which the GroEL/GroES system actually participates in the folding of newly-synthesized proteins in the cell is less defined and remains a subject for further studies. Ingenious biochemical and genetic approaches will be necessary to show whether our current view of chaperonin action indeed accurately reflects its modus operandi inside a living cell. KEY WORDS: protein folding, molecular chaperone, chaperonin, GroEL, GroES |
Abbreviation: DHFR) dihydrofolate reductase.
Although the basic principle remains unchallenged that the primary
sequence of a protein is sufficient to specify its three-dimensional
structure [1], it has become clear that protein
folding in the cell depends on helper proteins, of which a subgroup has
been collectively classified as molecular chaperones [2-4]. By recognizing unfolded or
partially denatured proteins, the predominant role of chaperones seems
to consist in the prevention of incorrect intra- and intermolecular
associations of polypeptide chains that would result in their
aggregation. The term "chaperonins" was initially coined for
the members of the Hsp60 family of molecular chaperones, which are
found in mitochondria, chloroplasts, and the eubacterial cytosol [5]. They are oligomeric complexes, composed of two
stacked rings, each of seven ~60-kD subunits that give shape to a
hollow cylinder [6, 7].
Unfolded polypeptides can bind at the inside of this cylinder.
Nucleotide binding to chaperonin subunits also allows binding of a
smaller cofactor to the chaperonin, which results in encapsulation of
the bound substrate protein in the cavity of the cylinder. ATP
hydrolysis is then typically required to promote substrate protein
release into the cavity, followed by folding to the native state. The
process of acquiring native or native-like tertiary structure in the
inside of a chaperonin complex becomes thus physically separated from
the event of protein synthesis in the cytosol. The recent elucidation
of X-ray crystallographic structures of high-molecular-weight
chaperonin complexes, the conformational analysis of chaperonin-bound
protein folding intermediates by NMR and mass-spectrometry, and results
from a series of sophisticated biochemical experiments now offer
detailed insight into the molecular mechanism of chaperonin action. In
this review, I shall discuss how chaperonins accomplish their task of
mediating productive protein folding in the cytosol, focusing on the
Escherichia coli chaperonin GroEL and its cofactor GroES.
STRUCTURE OF THE CHAPERONIN SYSTEM GroEL/GroES
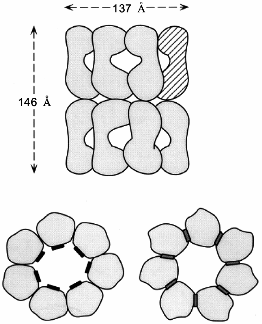
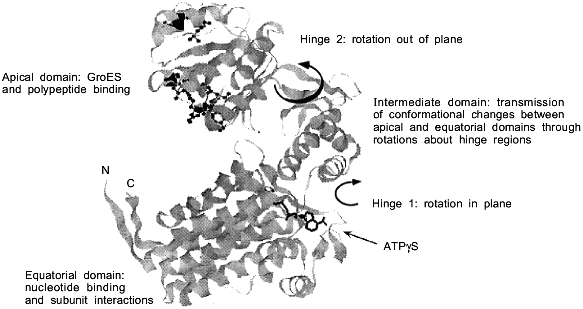
Two symmetrically stacked heptameric rings of identical 57-kD subunits form the GroEL cylinder (Fig. 1a). Its dimensions are 137 Å in diameter and 146 Å in height [8]. Each GroEL subunit (547 residues) consists of an apical domain (residues 191-376) that forms the opening of the cylinder and exposes a number of hydrophobic amino acid residues towards the center which are thought to interact with complementary surfaces of the non-native polypeptide substrate (Fig. 1b) [8, 9]. In the high-resolution X-ray structure of GroEL these residues are structurally ill-defined, indicating that they are part of flexible regions, a feature that probably makes them particularly suitable for recognizing binding motifs in a large variety of protein folding intermediates. A smaller intermediate domain (residues 134-190 and 377-408) joins the apical domain with the large equatorial domain, comprising the N-terminal part of GroEL up to residue 133, and amino acids 409-523. This domain contains the nucleotide-binding site (Fig. 1b) [10]. The Mg2+/K+-dependent ATPase activity of GroEL is characterized by positive cooperativity within one ring, and negative cooperativity between the two rings [11-14]. The structural features of the vicinity of the nucleotide binding site have been revealed by a crystal structure of GroEL with bound ATPgammaS [10]. In addition to binding nucleotide, the equatorial domain provides most of the intratoroidal side-to-side contacts and all the contacts between the two rings.
The C-termini of the GroEL subunits, which are flexible Gly-Met rich segments, are not crystallographically resolved, but all available evidence indicates that they protrude into the central channel at the level of the equatorial domains. This has implications for the mechanism of protein folding which occurs inside the cavity: transfer of an enclosed polypeptide between the two stacked GroEL-rings is prohibited. The channel itself has a uniform diameter of 45 Å, which expands at the level of the intermediate domains outward to ~90 Å. Assuming a certain flexibility of the apical domain, this would allow binding of substrate proteins up to a size of 50-55 kD. One GroEL tetradecamer binds typically only one substrate protein [15]. As will be discussed in detail below, the dimensions of the cavity change and increase considerably upon binding of the cofactor GroES on top of one GroEL ring.a Fig. 1. Structure of the GroEL chaperonin complex. a) Top: side view of the GroEL double ring cylinder. Bottom: top view of the nucleotide free form of GroEL (left), with polypeptide binding sites (in black) facing the cavity; in the ATP-bound form (right), polypeptide binding sites (dark gray) have been moved away from the central channel towards the intersubunit interfaces. b) Ribbon diagram of the GroEL subunit that is shown hatched in the diagram in Fig. 1a. In the apical domain, residues that have been shown to be involved in binding of unfolded polypeptide are highlighted. They include the hydrophobic residues Tyr-199, Ser-201, Tyr-203, Phe-204, Leu-234, 237, 259, Val-263, 264. The diagram was constructed from the coordinate file pdb1der.ent [10] using Rasmol.b
GroES is a heptamer of 10-kD subunits [16]. The two crystal structures of bacterial GroES complexes that have been solved show the protein as a flexible, dome-shaped structure composed of beta-sheets [17, 18]. The GroES ring measures 75 Å in diameter and 30 Å in height. Flexible-loop regions (residues 16-33) protrude from each subunit at the base of the dome. These mobile loop regions are critical for the binding of GroES to GroEL at the level of the apical domain [19, 20]. NMR-studies have demonstrated that upon binding the loop region adopts a different beta-hairpin conformation [19]. The binding sites for GroES and unfolded polypeptide on GroEL do partially overlap [8], but initial contact between GroES and GroEL is most likely made at residues in the chaperonin that are positioned more distant from the peptide binding site. In the process of establishing a tighter contact with GroEL, the smaller cofactor then competes with the bound substrate protein for the common binding sites in the chaperonin. This mechanism helps to eventually displace the substrate protein into the cavity for folding (see below).
SUBSTRATE RECOGNITION AND CONFORMATION OF GroEL-BOUND POLYPEPTIDES
What is the conformation of a (poly)peptide when it is bound to GroEL, and what exactly is recognized by the chaperonin? Mass spectrometry or NMR coupled with hydrogen-exchange techniques have been employed to analyze the structural properties of GroEL-bound substrate proteins [21-26]. In summary, GroEL-bound polypeptides fall into the range of collapsed, molten globule-like folding intermediates that are generated very early in folding. They are characterized by the presence of more or less stable native-like secondary structure in the absence of persistent tertiary interactions. These intermediates expose hydrophobic surfaces, as indicated by their ability to bind the hydrophobic fluorescent dye anilino-naphthalene sulfonic acid and by their distinct Trp-fluorescence spectra. Chaperonins can bind with high affinity to hydrophobic regions of such molten globule forms [15, 27]. As detected by mass spectrometry, hydrogen exchange kinetics of a GroEL-bound alpha-lactalbumin form show that it has the same degree of protection as the free molten globule form [21]. While studies of the NMR structure of GroEL-bound cyclophilin [22] came to the conclusion that secondary structure is for the most part absent, similar NMR/hydrogen exchange protection studies with beta-lactamase [24] and dihydrofolate reductase (DHFR) [25, 26] demonstrated that the bound proteins have conformations that are highly protected and may possess significant amounts of native-like structure. The particular experimental conditions, and, most importantly, the specific features of the tested model substrates may account for these apparently different results. Alternatively, these findings may simply reflect the ability of GroEL to bind polypeptides in various conformational states that occur along their folding pathways. Further insight into the nature of substrate recognition by GroEL has come from the recently solved crystal structure of a GroEL fragment that comprises the polypeptide binding site [28]. Remarkably, in this structure the extended amino-terminal tail of one GroEL fragment molecule was found to bind in the active site of a neighboring molecule, thus potentially mimicking the binding of an authentic peptide substrate. Seven residues are bound to the GroEL fragment in a rather extended conformation, making contacts through hydrophobic interactions and hydrogen bonds, that are formed between the main chain of the tail and side chains of the binding site.
Even subtle differences in the sequences of homologous proteins may be critical in determining their affinity for GroEL. For example, a comparison of the interaction of E. coli and murine DHFR with GroEL reveals that distinct loop regions in the eukaryotic protein may be primarily responsible for causing binding to the chaperonin [29]. E. coli DHFR, in which these loops are absent, does not bind to GroEL. Different chaperonins, on the other hand, may vary in their affinity for a given polypeptide. For example, yeast mitochondrial Hsp60, a GroEL homolog, binds only weakly to murine and human DHFR, which are very good substrates for GroEL and Neurospora crassa Hsp60 [15, 30, 31]. As already mentioned, the stoichiometry of binding is one substrate protein per GroEL complex. Negative cooperativity prevents binding of a second polypeptide at the opposite GroEL ring. However, this situation would arise only with a GroEL in a nucleotide-free state. As will be discussed in the next section, the chaperonin complex that binds a polypeptide to initiate a typical folding cycle is in the ADP state and has GroES bound to one end, so that only one GroEL ring is free and available for interaction with unfolded protein.
MECHANISM OF CHAPERONIN-ASSISTED PROTEIN FOLDING
The intriguing ability of chaperonins to mediate the folding of proteins that without their assistance would aggregate, has spurred on scientists involved in the analysis of this group of proteins to come up with a variety of models that could explain their mode of action. In the last two years, experimental evidence has provided strong and solid support for a mechanism whereby protein folding occurs in association with chaperonins, inside their cylindrical cavity [32-34]. It is now established that three features form the basis for successful chaperonin action: binding of unfolded polypeptides prevents aggregation; release of these proteins into the cylindrical cavity permits their folding in a shielded environment; and repeated cycles of release and rebinding of proteins, without intermittent transfer into the bulk solution, enable structural rearrangements of certain kinetically trapped protein species. In the following section, the individual steps of a such a complete GroEL/GroES reaction cycle will be described.
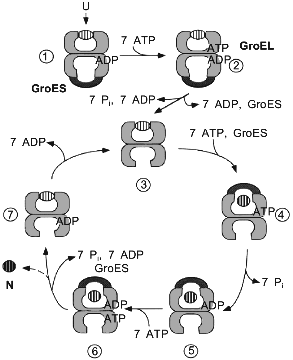
In contrast to many in vitro experiments, where unfolded proteins are bound to nucleotide-free GroEL, the chaperonin form that is encountered by substrate proteins in the cell is most likely a GroEL/GroES/ADP complex. First, unfolded polypeptide binds to such an asymmetrical GroEL/GroES complex in the trans-ring (the one not occupied by GroES) (Fig. 2, step 1). This event in itself, together with the binding and hydrolysis of ATP in the trans-ring (step 2), causes the dissociation of GroES and ADP (step 2 to 3). The nucleotide-free form of GroEL, with substrate protein bound (step 3), is only a short-lived intermediate. GroES, together with ATP, rapidly rebinds to either GroEL ring, apparently with equal chance. However, only binding to the polypeptide-containing ring leads to productive folding (step 4). At this point, GroES encloses the polypeptide in the GroEL cavity where it folds during the time of ATP hydrolysis as the wall of the cavity changes from hydrophobic to hydrophilic (see below) [35]. A single folding cycle ends after 15-20 sec when ATP is hydrolyzed in the trans-ring, resulting in the release of ADP and GroES from the polypeptide-containing cis-ring (steps 6 to 7). Folded protein leaves rapidly by diffusion upon the opening of the cage, while incompletely folded polypeptide can be recaptured as the hydrophobic binding sites of GroEL become available (step 7). This step, however, is also characterized by the release of a fraction of the total GroEL-bound protein in a non-native state [36, 37]. The extent to which it occurs in vivo is unknown, but some leakage may be necessary to clear GroEL of aberrant polypeptides that are unable to fold.
A central tenet of the chaperonin reaction cycle as we currently understand it is that protein folding occurs in the cylindrical cavity of GroEL, shielded from the cytosol, and that the substrate protein leaves the chaperonin in a more folded, less aggregation-prone conformation than its initially bound state. For subunits of oligomeric proteins this means that they are released as folded monomers for assembly with their partner subunits in solution [38]. Folding in association with the chaperonin is a GroES-dependent process. It is the binding of the cofactor that closes the cavity and prevents premature release of unfolded polypeptide into the external solution. In vitro, a number of proteins have been shown capable of folding with GroEL even in the absence of GroES. In this non-physiological situation, the protein would then rather leave the cavity in an unfolded form (release being caused by ATP hydrolysis) and fold in the bulk solution. Consequently, this reaction can be observed only with proteins that have the potential to reach the native state spontaneously under typical in vitro folding conditions. However, in the presence of GroES also these proteins are forced to fold in the cavity until they have reached a native-like or fully native conformation.Fig. 2. Model for the reaction cycle of GroEL/GroES-mediated protein folding. The chaperonin proteins are shown as vertical cuts through the rings. Individual steps of the cycle, leading from unfolded (U) to native (N) protein, are described in the text.
Most GroEL substrates require probably more than just one reaction cycle to reach that state [15]. (Re)binding of incompletely folded or misfolded polypeptide by GroEL is thought to provide a proof-reading function by allowing the structural rearrangement of unproductive intermediates, returning them to a productive folding pathway within the GroEL cavity. The energy of rebinding to GroEL would be used to overcome kinetic traps on the folding pathway before the major transition state is reached. There is still uncertainty, however, whether an active unfolding reaction takes place at the chaperonin surface. Data obtained from experiments concerning GroEL-induced unfolding of RNase T1 indicate that the chaperonin does not change the microscopic rate of unfolding in this case. Instead, it preferentially seems to bind the unfolded form of the protein, and, by thermodynamic coupling, shifts the equilibrium toward the unfolded state [39].
The structural rearrangement of unproductive folding intermediates by GroEL is apparently not mediated directly by the energy of cooperative ATP hydrolysis, but rather utilizes the binding energy of the hydrophobic interaction between polypeptide substrate and the apical GroEL domains. The main function of ATP hydrolysis is probably to optimize the rate of binding and release of GroES, thus coordinating the closing and opening of the GroEL cavity.
Each round of GroES binding and release is associated with the folding of the same fraction of GroEL-bound polypeptide, the efficiency of folding being dependent on the specific polypeptide. For rhodanese, a monomeric 33-kD protein that requires the complete GroEL/GroES system for folding, ~5% of the molecules fold fast enough to complete folding within the time window between GroES binding and release, which is governed by the GroEL ATPase [33]. On average a rhodanese molecule undergoes seven reaction cycles at GroEL before it has reached a stable native-like structure [15].
CONFORMATIONAL CHANGES IN THE CHAPERONIN DURING THE REACTION
CYCLE
GroES not only functions as a lid of the GroEL cavity in order to prevent premature release of substrate protein. GroES binding also causes massive conformational changes in the domains in the GroEL-ring it associates with. The apical domains in this ring move upwards and outward relative to the intermediate hinge domains (Fig. 1b) [35]. As a result, the volume of the central GroEL chamber in the associated ring increases significantly, forming the cage in which protein folding proceeds. The now dome-shaped cavity, with dimensions of 70 Å in height and diameter, is spacious enough to allow the enclosed proteins to explore their folding pathways without direct interference from the chaperonin itself. Equally important, the intermediate segments allow a hinge-like opening and twisting of the apical domains about the domain junctions, which occurs upon nucleotide binding in the equatorial domains. These movements are considerable and have been elegantly documented in a series of three-dimensional reconstructions of the complex from cryoelectron microscopic images [35]. The affinity of GroEL for unfolded polypeptide is strongly reduced because of a rotation of the apical domains about the hinge between the apical and intermediate domains, which occludes most of the hydrophobic-binding regions in the intersubunit interface (Fig. 1). This domain rotation is most pronounced in the ATP state, but to some degree takes place also in the ADP state. Finally, as the binding sites of the apical GroEL domains for GroES may overlap partially with those for the unfolded polypeptide, GroES binding would complete the burial of the hydrophobic patches of GroEL that bind unfolded polypeptide.
Nucleotide binding, even in the absence of GroES, is known to induce considerable conformational changes that extend beyond the apical domain into the opposite GroEL-ring [40]. As mentioned above, ATP binding occurs with positive cooperativity within one ring and displays negative cooperativity between the two rings. These changes are already sufficient to result in withdrawal of polypeptide binding sites and the dissociation of substrate proteins from GroEL in the absence of GroES [15]. Surprisingly, larger conformational changes are not evident in the recently solved crystal structure of GroEL with bound ATPgammaS [10]. The reason for that may be the use of a GroEL mutant for the crystallization that has lost the negative cooperativity between the two chaperonin rings [41].
CHAPERONIN ACTION AND FOLDING PATHWAYS in vivo
Most of the above described data on the GroEL/GroES chaperonin system have been derived from in vitro reconstitution studies with purified proteins. However, as the ultimate goal of all these studies is to understand protein folding as it occurs in the cell, the question is how relevant are these data are for chaperonin-assisted protein folding in vivo? Do they accurately reflect the way GroEL/GroES operates in the cell? Rather few studies have tried to address this issue, largely because of the technical difficulties in dissecting the dynamic protein folding process without unduly interfering with the reaction itself.
A hitherto neglected factor in our considerations of chaperonin-assisted protein folding is the unique physico-chemical environment in the cytosol that folding proteins are facing. The concentration of nascent (ribosome-bound) chains in the cytosol of E. coli is as high as ~35 µM, assuming a uniform distribution [42]. Their effective concentration will be significantly greater, however, because of molecular crowding. This term refers to the fact that a considerable fraction of the cellular volume is occupied by protein and other macromolecules at a total concentration of ~340 g/liter and is therefore not available to other macromolecules [43, 44]. Crowding is predicted to cause an increase in macromolecular association constants over those in dilute solution by several orders of magnitude. In contrast to in vitro conditions, where some of the GroEL-bound substrates are released in a non-native conformation, this apparent leakage of the chaperonin system is strongly reduced when folding is done in the presence of crowding agents [45]. Neither kinetics nor yields of folding are negatively affected by these conditions. The conclusion is that intermittent release of unfolded polypeptide into the bulk solution is not required for GroEL/GroES-mediated folding.
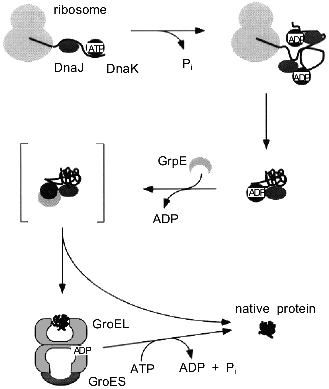
A second difference to in vitro GroEL/GroES-assisted folding reactions is the presence of additional chaperone components in the cell, especially members of the Hsp70 family (see [3, 4] for review). Evidence has been presented that DnaK, which is the E. coli Hsp70 homolog, and the chaperonins can act sequentially in mediating the folding of newly synthesized polypeptides in the eubacterial cytosol (Fig. 3) [46, 47]. In this folding pathway, DnaK and its co-chaperone, DnaJ, are thought to bind co-translationally to nascent chains on polysomes, thus preventing misfolding and aggregation [48, 49]. GroEL receives newly synthesized polypeptides only post-translationally for folding to the native state. Only a subset of all cytosolic proteins seems to depend on this pathway, on the basis of the cytosolic concentration of GroEL (3-5 µM) which is much lower than that of ribosomes and nascent chains (see above), and of the rate of synthesis relative to the rate of chaperonin-mediated folding measured in vitro. As for newly-synthesized proteins, it is estimated that GroEL/GroES is used by 5-10% of them, corresponding to ~200-300 different E. coli proteins in a size range of 20-50 kD [42, 50]. Whether a protein requires GroEL assistance or not may depend in part on the duration and the extent of the exposure of hydrophobic surfaces to solvent during folding. Furthermore, smaller proteins are less likely to require GroEL, because of faster folding kinetics and lower affinity to the chaperonin. In vitro, they may be able to bind, but often reversibly, and are known to dissociate even in the absence of ATP [51, 52]. The presence of seven binding GroEL subunits in a ring, each of which alone has a rather low affinity for peptides, may establish a threshold for stable binding by selecting for proteins of an adequate size that expose multiple binding elements. These proteins would be able to establish contacts with more than one subunit. On the other hand, advanced folding intermediates or native proteins that expose only one or two weak binding sites for GroEL would then be protected from undesired binding to the chaperonin.
As the chaperonin system has the ability to release proteins in an advanced folding state, it adds an element of directionality to the cellular protein folding pathway, where proteins are transferred from DnaK to GroEL. In vitro, the reverse reaction is also possible [53] if a protein cannot be folded by GroEL, for example because of a mutation. It is unknown whether such retrograde transfer from GroEL to DnaK occurs in significant amounts in vivo. Regardless of the role of DnaK in this process, a "built-in" leakage mechanism of GroEL/GroES may allow permanently damaged proteins that cannot adopt a stable conformation to leave the chaperonin and to interact with the degradative machinery. In fact, GroEL has been shown to be important for the proper degradation of cellular proteins [54].Fig. 3. Pathway of protein folding in the bacterial cytosol. Nascent polypeptide chains are bound by the molecular chaperones DnaJ and DnaK. A ternary complex is formed, consisting of nascent chain, DnaJ, and DnaK in its high-affinity ADP-state. Binding of GrpE to DnaK results in dissociation of the complex and release of substrate protein into solution. Single or multiple rounds of interaction of the folding protein with the DnaK/DnaJ/GrpE system lead to the native state. A subset of folding intermediates, 5-10% of all newly-synthesized cytosolic proteins, requires further assistance and is transferred to the GroEL/GroES system to complete folding.
CONCLUDING REMARKS
While the chaperonins have served as paradigms of cellular folding assistants over the last ten years, a variety of other chaperones with diverse structures and functions have been discovered since then and entered the limelight. Given the already detailed picture of the GroEL/GroES chaperonin that we have, what does future research on this system still have to offer? After mechanistic questions have been largely answered and structures for the components are on hand, the attention is expected to shift to questions of chaperonin function in vivo. Why does GroEL have two rings? Is it only to guarantee binding of substrate proteins in the presence of stable GroEL/GroES complexes? Is there any defined role for symmetrical GroEL/GroES complexes in which GroES is bound to each GroEL ring? Although it is clear that these forms, that can be generated in vitro under rather unphysiological conditions, are not required for a functional reaction cycle, do they improve the system in vivo? What are the authentic substrates for GroEL in the cell? How do larger proteins fold that do not fit into the GroEL cylinder cavity? And why does GroEL not interact with nascent polypeptide chains at ribosomes, but only post-translationally? Like in the past, research on chaperonins is likely to provide interesting results on cellular protein folding also in the coming years.
REFERENCES
1.Anfinsen, C. B. (1973) Science, 181,
223-230.
2.Ellis, J. (1987) Nature, 328,
378-379.
3.Hartl, F. U. (1996) Nature, 381,
571-580.
4.Martin, J., and Hartl, F. U. (1997) Curr. Opin.
Struct. Biol., 7, 41-52.
5.Hemmingsen, S. M., Woolford, C., van der Vies, S.,
Tilly, K., Dennis, D. T., Georgopoulos, C. P., Hendrix, R. W., and
Ellis, R. J. (1988) Nature, 333, 330-334.
6.Hohn, T., Hohn, B., Engel, A., Wurtz, M., and
Smith, P. R. (1979) J. Mol. Biol., 129, 359-373.
7.Hendrix, R. W. (1979) J. Mol. Biol.,
129, 375-392.
8.Braig, K., Otwinowski, Z., Hegde, R., Boisvert, D.
C., Joachimiak, A., Horwich, A. L., and Sigler, P. B. (1994)
Nature, 371, 578-586.
9.Fenton, W. A., Kashi, Y., Furtak, K., and Horwich,
A. L. (1994) Nature, 371, 614-619.
10.Boisvert, D. C., Wang, J. M., Otwinowski, Z.,
Horwich, A. L., and Sigler, P. B. (1996) Nature Struct. Biol.,
3, 170-177.
11.Bochkareva, E. S., Lissin, N. M., Flynn, G. C.,
Rothman, J. E., and Girshovich, A. S. (1992) J. Biol. Chem.,
267, 6796-6800.
12.Burston, S. G., Ranson, N. A., and Clarke, A. R.
(1995) J. Mol. Biol., 249, 138-152.
13.Gray, T. E., and Fersht, A. R. (1991) FEBS
Lett., 292, 254-258.
14.Todd, M. J., Viitanen, P. V., and Lorimer, G. H.
(1993) Biochemistry, 32, 8560-8567.
15.Martin, J., Langer, T., Boteva, R., Schramel, A.,
Horwich, A. L., and Hartl, F. U. (1991) Nature, 352,
36-42.
16.Chandrasekhar, G. N., Tilly, K., Woolford, C.,
Hendrix, R., and Georgopoulos, C. (1986) J. Biol. Chem.,
261, 12414-12419.
17.Hunt, J. F., Weaver, A. J., Landry, S. J.,
Gierasch, L., and Deisenhofer, J. (1996) Nature, 379,
37-42.
18.Mande, S. C., Mehra, V., Bloom, B., and Hol, W.
G. J. (1996) Science, 271, 203-207.
19.Landry, S. J., Zeilstra-Ryalls, J., Fayet, O.,
Georgopoulos, C., and Gierasch, L. M. (1993) Nature, 364,
255-258.
20.Landry, S. J., Taher, A., Georgopoulos, C., and
van der Vies, S. M. (1996) Proc. Natl. Acad. Sci. USA,
93, 11622-11627.
21.Robinson, C. V., Groß, M., Eyles, S. J.,
Ewbank, J. J., Mayhew, M., Hartl, F.-U., Dobson, C. M., and Radford, S.
E. (1995) Nature, 372, 646-651.
22.Zahn, R., Spitzfaden, C., Ottiger, M.,
Wüthrich, K., and Plückthun, A. (1994) Nature,
368, 261-265.
23.Zahn, R., Perrett, S., Stenberg, G., and Fersht,
A. R. (1996) Science, 271, 642-645.
24.Gervasoni, P., Staudenmann, W., James, P.,
Gehrig, P., and Plückthun, A. (1996) Proc. Natl. Acad. Sci.
USA, 93, 12189-12194.
25.Goldberg, M. A., Zhang, J., Sondek, S., Matthews,
C. R., Fox, R. O., and Horwich, A. L. (1997) Proc. Natl. Acad. Sci.
USA, 94, 1080-1085.
26.Groß, M., Robinson, C. V., Mayhew, M.,
Hartl, F.-U., and Radford, S. E. (1996) Protein Sci., 5,
2506-2513.
27.Hayer-Hartl, M. K., Ewbank, J. J., Creighton, T.
E., and Hartl, F.-U. (1994) EMBO J., 13, 3192-3202.
28.Buckle, A. M., Zahn, R., and Fersht, A. R. (1997)
Proc. Natl. Acad. Sci. USA, 94, 3571-3575.
29.Clark, A. C., Hugo, E., and Frieden, C. (1996)
Biochemistry, 35, 5893-5901.
30.Rospert, S., Looser, R., Dubaquié, Y.,
Matouschek, A., Glick, B. S., and Schatz, G. (1996) EMBO J.,
15, 764-774.
31.Ostermann, J., Horwich, A. L., Neupert, W., and
Hartl, F.-U. (1989) Nature, 341, 125-130.
32.Martin, J., Mayhew, M., Langer, T., and Hartl,
F.-U. (1993) Nature, 366, 228-233.
33.Mayhew, M., da Silva, A. C. R., Martin, J.,
Erdjument-Bromage, H., Tempst, P., and Hartl, F.-U. (1996)
Nature, 379, 420-426.
34.Weissman, J. S., Rye, H. S., Fenton, W. A.,
Beechem, J. M., and Horwich, A. L. (1996) Cell, 84,
481-490.
35.Roseman, A. M., Chen, S., White, H., Braig, K.,
and Saibil, H. R. (1996) Cell, 87, 241-251.
36.Weissman, J. S., Kashi, Y., Fenton, W. A., and
Horwich, A. L. (1994) Cell, 78, 693-702.
37.Todd, M. J., Viitanen, P. V., and Lorimer, G. H.
(1994) Science, 265, 659-666.
38.Zheng, X., Rosenberg, L. E., Kalousek, F., and
Fenton, W. A. (1993) J. Biol. Chem., 268, 7489-7493.
39.Walter, S., Lorimer, G. H., and Schmid, F. X.
(1996) Proc. Natl. Acad. Sci. USA, 93, 9425-9430.
40.Langer, T., Pfeifer, G., Martin, J., Baumeister,
W., and Hartl, F.-U. (1992) EMBO J., 11, 4757-4765.
41.Aharoni, A., and Horovitz, A. (1996) J. Mol.
Biol., 258, 732-735.
42.Ellis, R. J., and Hartl, F.-U. (1996) FASEB
J., 10, 20-26.
43.Zimmerman, S. B., and Trach, S. O. (1991) J.
Mol. Biol., 222, 599-620.
44.Zimmerman, S. B., and Minton, A. P. (1993)
Annu. Rev. Biophys. Biomol. Struct., 22, 27-65.
45.Martin, J., and Hartl, F.-U. (1997) Proc.
Natl. Acad. Sci. USA, 94, 1107-1112.
46.Langer, T., Lu, C., Echols, H., Flanagan, J.,
Hayer, M. K., and Hartl, F.-U. (1992) Nature, 356,
683-689.
47.Gaitanaris, G. A., Vysokanov, A., Hung, G.-C.,
Gottesman, M. E., and Gragerov, A. (1994) Mol. Microbiol.,
14, 861-869.
48.Hendrick, J. P., Langer, T., Davis, T. A., Hartl,
F.-U., and Wiedmann, M. (1993) Proc. Natl. Acad. Sci. USA,
90, 10216-10220.
49.Kudlicki, W., Odom, O. W., Kramer, G., and
Hardesty, B. (1994) J. Mol. Biol., 244, 319-331.
50.Horwich, A. L., Low, K. B., Fenton, W. A.,
Hirshfield, I. N., and Furtak, K. (1993) Cell, 74,
909-917.
51.Corrales, F. J., and Fersht, A. R. (1995)
Proc. Natl. Acad. Sci. USA, 92, 5326-5330.
52.Gray, T. E., Eder, J., Bycroft, M., Day, A. G.,
and Fersht, A. R. (1993) EMBO J., 12, 4145-4150.
53.Buchberger, A., Schröder, H., Hesterkamp,
T., Schönfeld, J. J., and Bukau, B. (1996) J. Mol. Biol.,
261, 328-333.
54.Strauss, D. B., Walter, W. A., and Gross, C. A.
(1988) Genes Dev., 2, 1851-1858.