REVIEW: Competition Involving Biogenic NO
A. A. Nedospasov
Institute of Molecular Genetics, Russian Academy of Sciences, pl. Kurchatova, Moscow, 123182 Russia; fax: (095) 196-0221; E-mail: nedospas@img.ras.ru
Received July 1, 1997
This review considers competitive reactions of NO biosynthesis and its interaction with its targets including enzyme competition for arginine, competition between the substrates in the NO-synthase active sites, competitive sources of arginine, multiple states of NO-synthases resulting from competition of reducible cofactors (FAD, FMN, BH4, heme), competition between aqueous and lipophilic phases for NO resulting in efficient micellar catalysis, and other aspects of competition involving biogenic NO.
KEY WORDS: nitric oxide, NO-synthase, competitive substrates, peroxynitrite, S-nitrosothiols, arginine, tetrahydrobiopterin
Abbreviations: AA) arachidonic acid; AL) arginosuccinate lyase; Arg) arginine; AS) arginosuccinate synthetase; BH4) tetrahydrobiopterin; COG) cyclooxygenase; EDRF) endothelium-derived relaxing factor (prior to 1987, unidentified compound activating guanylate cyclase; after 1987, NO or its equivalent); GC) guanylate cyclase; GSNO) S-nitrosoglutathione; LHRH) luteinizing hormone releasing hormone; NHA) Nomega-hydroxyarginine; NMA) Nomega-methylarginine; NOS) NO-synthase; PC1, PC2, KEX) arginine-specific processing proteases (propeptide-convertases); Y+) transport system for positively charged amino acids.
In biochemical publications, the term "competition" has many
diverse meanings and is used to describe phenomena when there is a
choice between several alternative objects (compounds, reactions, or
processes) that are usually characterized by mutual negative feedback,
i.e., less of one versus more of another. Herein, the term
"competitive enzymes" corresponds to enzymes with at least
one common substrate; for example, arginase, arginyl-tRNA-synthetase,
and NO-synthase have one common substrate, arginine. "Competitive
sources" are substances, reactions, or processes generating the
same product. For example, arginyl aminopeptidase, Y+
transport system, and arginosuccinate lyase (AL) can be considered as
competitive sources of cellular arginine, at least when they are
inhibited by arginine excess. "Competitive substrates" are
substrates undergoing conversion in the very same enzyme active site
via a similar mechanism. For example, arginine and homoarginine are
competitive substrates of NO-synthase (both are oxidized via similar
mechanism generating NO, water, and citrulline or homocitrulline,
respectively). "Competitive targets" are compounds reacting
with NO.
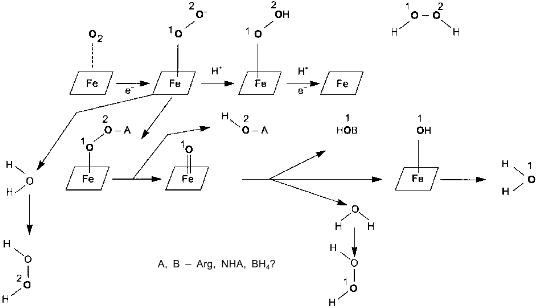
The discovery of biogenic NO and understanding of its role in higher organisms is one of the most unexpected and brilliant achievements of biochemistry. The enzymes producing the major part of NO, NO-synthases (NOS), are unique in the complexity of their organization and include a multitude of various cofactors (FMN, FAD, BH4, heme, calcium--calmodulin, and some scientists think that glutathione is also a cofactor) and at least three very dissimilar substrates (arginine, oxygen, and NADPH). Water is the donor of protons and thus a substrate but it was considered only as a reaction product. The total equation of NOS-catalyzed reaction includes 5-electron (N3- --> N2+) oxidation of the arginine nitrogen atom coupled with NADPH oxidation:
Surprisingly, experimental studies of the sources of hydrogen atoms in the generated water molecules have not yet been performed. However, simple coefficients (2/3) in Eq. (1) are the serious problem for everyone who wants to understand the functioning of NOS; in case of electron transport from NADPH to heme via flavin cofactors, the fractional coefficient in the equation can have certain physicochemical explanation because flavins can undergo one-electron redox transformations forming semiquinones, but half of a proton does not make any sense. Hence, Eq. (1) implies that either both active sites of the dimeric NOS molecule are functionally interacting (for example, synchronously, in counterphase, or have completely different functions; then, doubling of equation coefficients is required) or there are at least two different cycles of activity and the enzyme can return to the initial state after an even number of turns.Fig. 1. NO biosynthesis. The upper part shows inactive enzyme in the absence of Ca2+. NADPH reduces flavins but the heme is not reduced. Arg and BH4 (on the left) bind to the active site but are not oxidized. The lower part shows that Ca2+ binding induces structural rearrangement and electron systems of flavins and heme are combined. Heme is reduced, binds O2, oxidizes arginine to N-hydroxyarginine (NHA) and then during the second cycle, to citrulline (Cit) and NO. (The drawing shows that arginine and NHA are oxidized at the same heme, but this is not known for sure. The role of tetrahydrobiopterin (BH4) in the catalysis is not shown.)
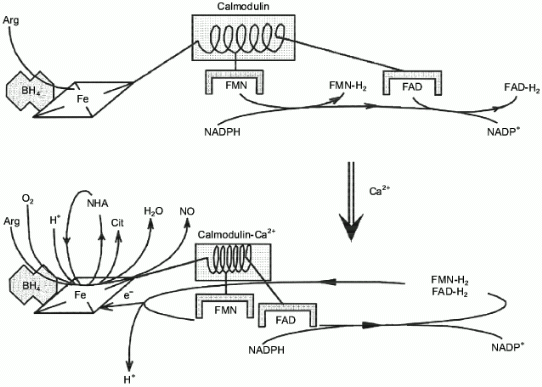
There are essentially no data confirming that oxygen binds to the same heme during the first and second oxidation stages and an intuitive assumption that two identical subunits should unequivocally form identical active sites is apparently wrong; recent results indicate that heterodimers have no catalytic activity if one of the subunits cannot bind heme [3]; thus, both hemes participate in catalysis.
The function of BH4 is poorly understood. Apart from an allosteric role in the formation of the oxidative active site, it is apparently required for the binding of the second atom of the first oxygen molecule (upon arginine oxidation into NHA one oxygen atom is incorporated into NHA and another one is released as a water molecule) but this process has not been studied in great detail. Taylor et al. and Gorren et al. in this issue [4, 5] discuss the structure and mechanism of regulation of NOS activity in detail and review the preceding publications (see [6] and references therein).
Discussion of NO biochemistry becomes more convenient when the metabolites are divided into two classes with odd or even number of electrons. Amino acids, sugars, nucleotides, and most of the other organic compounds are "even" (have an even number of electrons) and are diamagnetic. NO (15 electrons, paramagnetic) is of the second class. There is a small number of even paramagnetic compounds (biradicals containing two uncoupled electrons) including the common metabolite, oxygen (triplet dioxygen; O2).
Homolytic breakage of almost any chemical bond is associated with the formation of two odd molecule from one even:
NO reversibly reacts with metal ions forming complexes (evidently, outer electrons of NO migrate to vacant orbitals of the metal atoms). In s- and p-elements, such complexes are usually unstable and exist only at high concentrations of free NO but in case of transition metals, extremely stable complexes have been described. Tetranitrosyl chromium (Cr(NO)4) is an interesting example of the formally "zero valency" element; it is purified by sublimation; sodium nitroprusside (Na2[Fe(CN)5NO]) is crystallized from boiling water; it decomposes forming NO only at 400°C. High stability of such complexes can be due to the backbonding interaction; interaction of the pi-loosening orbital of NO with d-orbitals of the metal generates a new hybrid orbital where the metal electrons migrate (this is backbonding) (for details see review by Fukuto [7] and references therein).
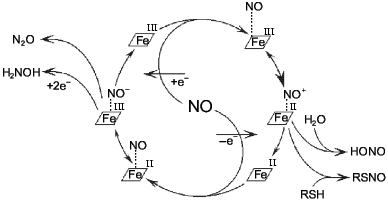
Most odd metabolites are transition metal ions and their complexes with organic ligands. Co, Cr, Cu, Fe, Mn, Mo, and V exist in sequential oxidation states and the energy of transition between these states is often small depending on the surrounding atoms (reactions associated with odd-even transition are possible at low temperatures). Formation of the metal--NO complex (independently from stability of the complex) can be associated with concomitant intramolecular redox reactions. For example, NO forms complexes with heme; in the case of Fe2+ the complex is extremely stable but oxidation into Fe3+ results in reversible complexation; apparently, the latter complex is an equilibrium structure with Fe2+ and Fe3+ (Fig. 2).
At low NO concentrations, the metal ion is often one-electron oxidizer or (less often) reducer, thus enabling the formation of an even product in a reaction with a common even metabolite. Hence, nitrite and thionitrites (S-nitrosothiols), which are even metabolites of nitrogen in the oxidation state +3, can be formed during one-electron oxidation of NO by a transition metal ion without utilizing another NO molecule. Similarly, the oxygen--hemoglobin complex forms nitrate and methemoglobin in reaction with NO:Fig. 2. NO--heme complexes as sources of NO+ and NO- ions. The NO+ ion is the nitrosylating agent and is detected indirectly by formation of thionitrites (in the presence of thiols) or nitrite. Nitrous oxide (N2O) and hydroxylamine are competitive products of conversion of NO--heme (Fe2+) complex.
At low physiological concentrations of NO and its main potential targets, transition metal ion complexes, NO can react at a rather large distance from the site of its formation because of its high diffusion coefficient, good membrane permeability and solubility in water and especially in lipids. Lancaster suggested that NO diffuses according to the second law of Fick and its utilization is a first-order reaction for NO; considering the known half-life of NO in the body (5-30 sec), he estimated the distance corresponding to two-fold decrease in NO concentration to be 160 nm (8 cell diameters) [9]. Considering the non-homogeneity of the medium and active transport, this distance can significantly differ from this value; for details see below. Thus, the number of potential targets competing for reactions with NO can be extremely high (transition metal complexes in at least a thousand cells adjacent to the NO source).
Hence, competition is a common feature of biogenic NO including competition of enzymes for the substrates and cofactors during its synthesis to competition of the targets that inactivate NO during manifestation of its biological activity.
COMPETITIVE ENZYMES
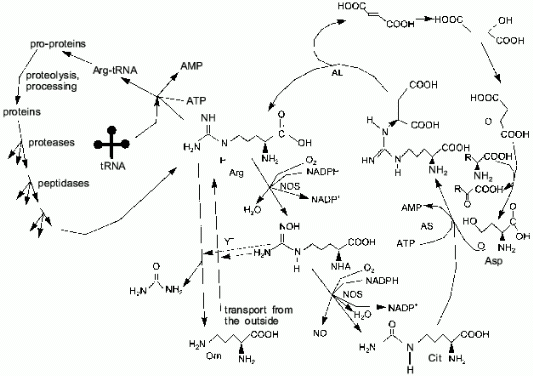
One of the terminal guanidine group atoms of arginine is the source of nitrogen for NO synthesis by NOS. In most cell types, the urea cycle is not complete and arginine cannot be synthesized (in humans, arginine is synthesized in the liver); thus, most cells depend on arginine import. There are three sources of arginine: transport (Y+ transport system), proteolysis, and citrulline amination (arginosuccinate synthetase (AS) and arginosuccinate lyase (AL) are expressed not only in the liver as it was thought earlier, but to a certain extent in virtually all cell types [10]) (Fig. 3). Macrophages and similar cells produce high amounts of NO and they might be thought to rapidly utilize all free arginine, converting it to citrulline. Indeed, this does not happen because AS and AL synthesis is induced in macrophages simultaneously with NOS; thus, arginine is resynthesized from citrulline and aspartic acid. Nitrogen of other amino acids can be oxidized to NO due to transaminases [11-15]. It was shown [16] that synthesis of NOS is maximal 5 h, synthesis of AS is maximal 6-12 h, and synthesis of AL is maximal 24 h after induction; this is reasonable because, at first, NOS can utilize available arginine. If AL is constitutive and AS is absent prior to induction, the products of the first AS molecules can be cleaved by the initial AL.
Usually, a major part of arginine is consumed in protein synthesis. Apart from NO biosynthesis, arginine is cleaved by arginase, generating urea and ornithine and, thus, it is irreversibly lost for the cells which lack the complete urea cycle. Other arginine metabolism pathways are known (for example, decarboxylation) but their influence on the total pool is apparently low.Fig. 3. Arginine metabolism. The cycle on the right shows NO-synthase (NOS)-catalyzed conversion of arginine to N-hydroxyarginine (NHA) and then to citrulline. Arginosuccinate synthetase (AS) and arginosuccinate lyase (AL) can restore the guanidine group utilizing the NH2-group of aspartate. Due to transaminases (right), amino groups of other amino acids can be used to convert citrulline to arginine as well. The cycle on the left shows the utilization of arginine in protein synthesis, while proteolysis restores the arginine pool. Vertical arrows correspond to Y+ system-dependent transport of arginine inside the cell, whereas arginase cleaves arginine generating ornithine and urea. NHA inhibits both pathways (dashed lines).
A common substrate enables a self-regulating system even for two enzymes with Michaelis kinetics (Fig. 4a); if Km1 << Km2, at certain substrate concentrations when the rate of the first enzyme is proportional to [S], the rate of the second one does not depend on [S]. In case of NOS and arginase, kcat and Km are different by several orders of magnitude and kcat/Km for arginase is significantly higher (for example, in rat liver arginase kcat/Km = 2.6.106 M-1.sec-1 [17] but Km for arginine is over 1 mM; in NOS, kcat/Km varies depending on the enzyme species (Table 1) but it does not exceed 5.105 M-1.sec-1 with Km < 30 µM). Unlike competitive substrates (see below), competitive enzymes in general decrease the catalytic efficiency of each other only by changes in the substrate concentration. However, it was shown that NHA (an intermediate product of arginine oxidation to NO) is a potent inhibitor of arginase (Ki = 42 µM) [18]. It was suggested that this inhibition can be of regulatory importance since it would block the decomposition of arginine which is required for NO synthesis. On the other hand, it is unclear what are the in vivo conditions corresponding to such high NHA concentration that is sufficient for inhibition. Arginase contains at least six manganese ions per active trimer. Possible feedback mechanisms in the NOS--arginase system should include the interaction of these ions with NO. It is important that in macrophages, lipopolysaccharides induce NOS and arginine-cleaving arginase II, but interferon-gamma enhances NOS induction and blocks transcription of the arginase gene [42]. Thus, there are at least two programs of NO synthesis in macrophages: under moderate conditions, when removal of excessive arginine can be required, and under maximal conditions, when all arginine should be oxidized (in both cases, initial arginine is oxidized first).
Fig. 4. a, b) Effect of [S] on v for two enzymes (1, 2) competing for a common substrate at Km1 << Km2 and kcat1 << kcat2: a) kcat/Km1 > kcat/Km2; b) kcat/Km1 < kcat/Km2. Inserts show the enlarged initial segments of the curves. In all cases curve (3) corresponds to the ratio (1)/(2).
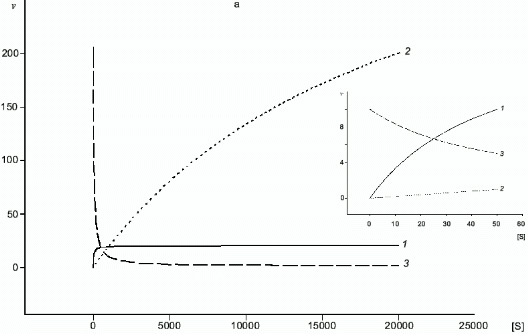
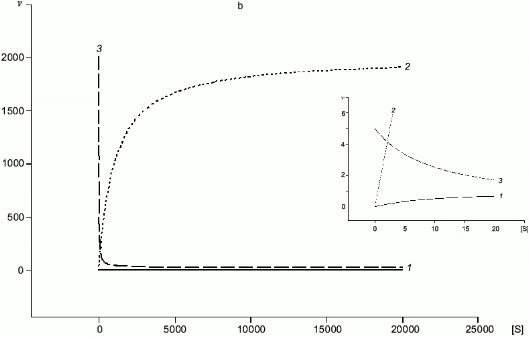
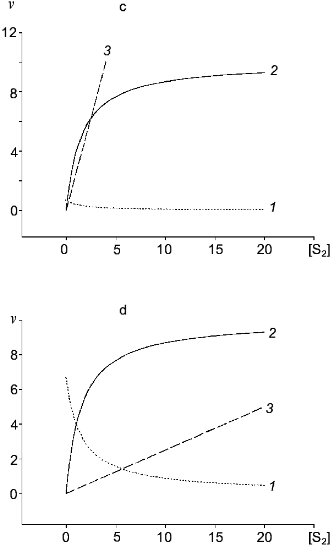
Table 1. Kinetic constants of NO-synthasesFig. 4. c, d) Effect of [S2] on v for two competitive substrates at constant [S1]; c) kcat1 << kcat2; d) kcat1 >> kcat2; curve (3) corresponds to vi/vj = [Si]kcatiKmj/[Sj]kcatjKmi and is linear in all cases.

|
Note: 1) Rat cerebellum; 2) mouse macrophages; 3) porcine cerebellum; 4) bovine brain; 5) rat macrophages; 6) human brain; 7) human hepatocytes; 8) rat liver; 9) bovine endothelium; 10) porcine endothelium; 11) bovine endothelial NOS expressed in E. coli. a Enzyme was isolated without thiols. b Dithiothreitol was added to enzyme isolated without thiols. c Without arginine. d With arginine. *Effect of BH4 concentration on v was measured. |
Two forms of NOS can behave in a similar way (differences in kinetic constants exceed one order of magnitude, see Table 1) because in many cell types, various NOS forms are expressed. Transgenic knockout mice were generated lacking each of the NOS isoforms. Unexpectedly, they were all viable (why such a complex enzyme system is required if it is not vitally important; see comments of Snyder [43]). Knockout of NOS-1 gene results in aggressiveness and excessive sexuality; knockout of NOS-2 results in enhanced sensitivity to infection; deletion of NOS-3 causes moderate increase in blood pressure [44-46]. It was suggested that the functions of deleted NOS are performed by the residual isoforms. Selective inhibition of NOS is important in various medical applications. Considering variable affinity to arginine, arginine analogs and inhibitors of parallel metabolic pathways can be very efficient.
To reach the maximal rate of an enzyme reaction, substrate concentration should be several-fold higher than Km. Indeed, arginine concentration in cells depends on tissue type and varies in a time-dependent manner. Assay of intracellular arginine concentration (0.1-2 mM) gives average values that can vary with time and subcellular localization. Hence, arginine concentration can regulate NOS activity in vivo.
Bune et al. have demonstrated that arginine concentration in rat plasma (225 µM) can be lowered to zero by addition of large amounts of arginase [47]. Arginine decomposition significantly decreases NO synthesis during endotoxic shock. This indicates that even though macrophages can resynthesize arginine from citrulline (NOS product), extracellular arginine is essential for NO synthesis.
Unlike NOS-2, two other isoforms are constitutively present in the cell but are active only at high calcium concentration in the medium. At least under certain circumstances, the efficiency of arginine resynthesis from citrulline is low. In platelets, it was shown that addition of L-arginine into the extracellular medium enhances NO synthesis [48]. Thus, arginine is not in excess and its concentration can regulate NOS activity. (Evidently, our ancestors had empirically discovered this before the times of the Bible; during religious abstinence, arginine consumption with food was significantly limited with the corresponding consequences.)
A group of French scientists [49] studies several types of NOS-expressing tumor cells; they have shown that in the presence of BH4 and NADPH (without flavins), NHA is the main NOS product. NHA accumulation inhibits arginase and the Y+ transport system, resulting in cytotoxic effects.
COMPETITIVE SUBSTRATES
Similar to catalytic oxidation of ammonia to NO, arginine oxidation to NO and citrulline is exothermic and does not need to be coupled with some other reactions to shift the equilibrium rightwards. NO biosynthesis by NOS is coupled to flavin-dependent oxidation of NADPH that is also exothermic and it is associated with generation of a highly reactive peroxide intermediate which can oxidize most biological molecules. For example, N-methylarginine (NMA) was first identified as a competitive inhibitor of NOS; detailed investigation revealed that it is the substrate and the final reaction products are NO and demethylated citrulline, i.e., the methyl group is oxidized as well [28]. Thus generated formaldehyde is apparently responsible for irreversible inactivation of the enzyme. Other natural arginine analogs, which can efficiently compete with it in NO-synthases forming NO, are not known (D-isomer and L-canavanine are not substrates; for details see [50]). It is not a big surprise that with an excess of NADPH without arginine and BH4, NADPH oxidation by NOS is associated with hydrogen peroxide generation, i.e., formally, water is oxidized [36, 51-53] (Fig. 5).
Formation of peroxide can be explained by at least three distinct mechanisms. First, dioxygen binds to the heme and is then reduced by electrons from flavins. In this case, both oxygen atoms of the generated H2O2 molecule come from O2 and only protons come from H2O. Alternatively, one atom of the initial O2 molecule is activated in the enzyme active site and attacks water, whereas another attacks a coupled substrate (BH4 or arginine?). Thus generated H2O2 molecule should have one oxygen atom from water and another from the O2 molecule. In this case, NOS can be considered as NADPH-oxidase where water and arginine (or BH4) are competitive co-substrates in the two-electron oxidation. Finally, if there is a coupling with arginine or BH4 oxidation, peroxide can contain two oxygen atoms coming from water. Although the experiments are evidently simple, they have not been performed in any NOS isoform and the real mechanism of peroxide formation is unknown.Fig. 5. Competition in the NOS active site. Depending on oxidation mechanism, various isotopically substituted products are formed.
In the case of competitive substrates, the ratio of conversion rates is calculated according to the equation:
Table 2. Expected according to Eq. (4) and actual effects of arginine and BH4
on H2O2 formation by NOS

NOS catalysis is not limited to competition for the oxidizer; artificial "substrates" like cytochrome c compete with heme for flavin reduction; apparently, dissociation of NO from the active site is also competitive (see below).
COMPETITIVE SOURCES
Constitutive NOS is activated by calcium ions and starts working in an arginine-independent manner. At low arginine and/or BH4 concentrations, H2O2 is a parallel enzyme product. Nonenzymic reaction between NO and superoxide anion is very efficient and results in the formation of highly toxic peroxynitrite that induces apoptosis (such a combination is used by macrophages to generate a cytotoxic compound); thus, it seems very unlikely that in vivo neuronal NOS could function at low arginine concentrations generating peroxide.
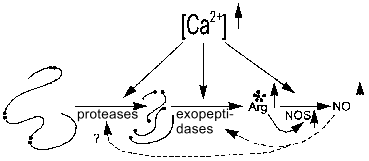
Most amino acids in the cell are stored as proteins. In those cells which cannot synthesize amino acids de novo, the pool of free amino acids is supplemented by proteolysis; the main consumers of amino acids are aminoacyl tRNA-synthetases of protein synthesis system (left cycle in Fig. 3). At a constant rate, these processes should not rapidly influence the concentrations of amino acids. When total rate of proteolysis increases, concentrations of all amino acids can slightly but transiently increase. However, if acceleration of proteolysis involves only the substrates enriched in a certain amino acid, increase in its concentration can be fast and significant. The Ca2+-dependent system of pro-protein processing of the regulatory secretory pathway meets these requirements.
The recognition sites of the regulatory processing proteases include single or more often groups of arginine residues. For example, furin and KEX-like proteases cleave the peptide chains of the secreted proteins at Arg-X-Y-Arg sites where Y is lysine or arginine and X is any amino acid [60, 61]. Then, exopeptidases cleave arginine residues at the terminal parts of the molecule. In fact, processing is the removal of several amino acids from the pro-protein and among these amino acids, arginine is predominant.
The activity of certain pro-protein processing proteases is not constant. PC1 is constitutively active and does not require any additional cofactors. On the contrary, PC2 is a calmodulin-dependent enzyme and requires elevated calcium concentrations. Arginine residues adjacent to the processing protease-cleaved bonds can supplement the free arginine pool in the presence of active exopeptidases. However, it was demonstrated that certain arginine exopeptidases are also calcium-dependent and are inactive under standard conditions; hence, arginine residues are not cleaved and are used for secretory granule formation [62, 63]. Calcium enhances the activities of Arg-specific processing proteases of the second type and of exopeptidases, thus causing release of free arginine. So, increase in arginine concentration is induced by the secretion signal but reaches its maximal values after a certain delay.
Neuropeptides are an important class of molecules participating in signal transduction between cells. They are synthesized as large precursor molecules containing several sequentially linked copies. Such pro-peptides are functionally inactive and can be stored in the cells until they become needed. The copies of neuropeptides in the precursor molecule are separated by the arginine-rich sites. Their processing is essentially similar, but because the precursor molecule contains several intermediate sites, the total number of arginine molecules formed during the conversion of the precursor molecule is proportionally higher than that generated during cleavage of high-molecular-weight proteins.
Increase in cellular arginine concentration induced by the formation of secreted proteins or regulatory peptides should influence the enzymes utilizing arginine. Enzymes with low Km for arginine (Km of aminoacyl tRNA-synthetases for arginine is below 10 µM [64]) would be poorly sensitive to such increased concentration even if [Arg] is increased several-fold because their active sites were already saturated at the initial arginine concentration. On the contrary, enzymes with lower arginine affinity would have optimal conditions for catalysis. In the presence of a competitive substrate, the conversion rates of both substrates are changed according to Eq. (4). Thus, in cells expressing NOS and secreting regulatory peptides, the activity of the former can be regulated by the synthesis of the latter. Such regulation (Fig. 6) depends on the initial and proteolysis-generated arginine and can maintain the optimal levels of NO and/or peroxynitrite synthesis as well as complete inhibition of peroxynitrite synthesis in cells where it should not be synthesized.
The activity of NOS and proteases of processing of regulatory peptides maintaining the arginine pool (including that for NOS) can be calcium-dependent or -independent; hence, several classes of Arg-specific hydrolases--arginine--NO systems can be considered. The effects of inhibition of these proteases and peptidases on NOS activity are needed to study such systems.Fig. 6. Increase in [Arg] and [NO] by Ca2+-dependent proteolysis. Increased calcium concentration activates Arg-specific processing proteases cleaving pro-peptide at Arg-rich sites (arginine residues shown with black dots), certain Arg-specific exopeptidases cleaving terminal arginine residues, and NOS (NOS-2 is active without Ca2+). Increased [Arg] induces enhanced synthesis of NO by NOS. Generated NO regulates amide hydrolases (dashed lines).
The mechanism requires significant increase in arginine concentration induced by Ca2+-dependent proteolysis. Rough estimate indicates that this can happen not only in cells simultaneously secreting NO and polypeptides but in adjacent cells when the final processing stages take place outside of the cell or when arginine is secreted together with secretory granules. The NO flux can be the signal triggering peptide secretion (luteinizing hormone releasing hormone [65, 66]; discussed below; Fig. 7) and Arg-specific hydrolases (reviewed in [63]); thus, chemical oscillatory systems similar to the Belousov--Zhabotinsky reaction can exist and these systems can be associated with oscillations in concentrations of NO, arginine, and secreted peptides. This hypothesis has not been tested experimentally, but it was shown that NO participates in regulation of short oscillations in the brain [67]. The possible oscillation frequencies for such a mechanism are also unknown.
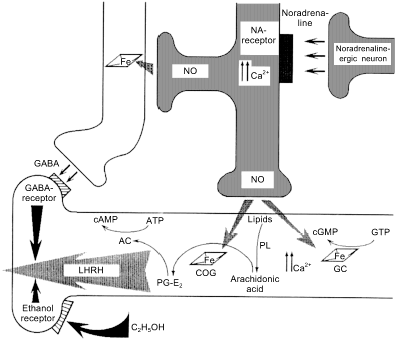
Fig. 7. Regulation of secretion of luteinizing hormone releasing hormone (LHRH). A NO-ergic neuron (center) generates NO fluxes and thus activates guanylate cyclase (GC) and cyclooxygenase (COG) of an LHRH-ergic neuron. This induces an increase in [Ca2+] and phospholipids are cleaved by phospholipase (PL) generating arachidonic acid that in converted into prostaglandin PG-E2 by COG. The prostaglandin activates adenylate cyclase and cAMP is the direct signal to start secretion. gamma-Aminobutyric acid (GABA)-ergic neuron (left) is activated by the NO flux derived from the same NO-ergic neuron and blocks secretion shortly after its onset. Ethanol receptor (left lower part) also blocks LHRH secretion.
COMPETITIVE COFACTORS
The combination of two different oxidation reactions (two-electron Arg --> NHA and three-electron NHA --> NO) in a single active site with independent regulation of each reaction is a serious problem of NOS enzymology. The second reaction is inhibited under FMN deficiency but unlike the first one, does not require BH4 and dimerization of the subunits.
The presence of FAD and FMN in each subunit is a unique feature of NOS. The NOS molecule can have many forms differing in oxidation states of each cofactor since it is dimeric and contains heme and BH4. These forms are apparently competitive in redox reactions and substrate binding. Moreover, Venema et al. [68] have shown that in neuronal and endothelial NOS, there are at least three types of dimeric molecules (according to the authors, if oxygenase and reductase domains can be considered as "head" (H) and "tail" (T) these forms correspond to HH, HT, and TT) depending on the type of domains participating in the formation of the secondary structure. In NOS-2, only HH domains have been demonstrated.
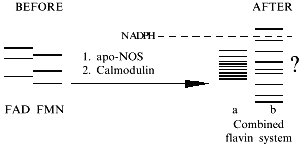
In NOS, flavins are the wick and sponge of electrons by accumulating them in the active sites of NADPH oxidation and transferring them to the hemes. It is important that NADPH supplies pairs of electrons which leave the flavin sponge one-by-one. Usually, in reviews, the electron transfer is considered as a sequence:
Scheme
Matsuoka et al. [21] compared NOS to cytochrome P450-reductase. Their interpretation of NOS electrochemistry includes changes in heme potential as the result of arginine binding, similar to that observed in cytochrome P450 upon camphor binding (decrease in E from -0.3 to -0.17 V). This assumption is supported by an increased rate of heme reduction in the presence of arginine.
It has been proven that two hemes of the NOS molecule are not equivalent, but the detailed reason for such a difference is unknown. Xie et al. [3] showed that the mutant NOS-2 proteins do not bind heme and have no NO-synthase activity, but they can form inactive heterodimer with the wild-type subunit. Thus, two hemes are required for NO synthesis from arginine, whereas NHA can be oxidized by a single subunit. However, whether this functional difference (in hemes or subunits) is fixed or is subject to change (randomly or non-randomly) during each cycle of activity is unknown. Unlike the macrophage enzyme, NOS-1 forms dimers even in the absence of arginine and BH4, but if there is no heme there are no dimers [70]. Moreover, addition of hemin to apo-NOS does not result in an active enzyme; the heme binds but activity does not appear. Heme-deficient enzyme cannot bind BH4 and arginine. Abu-Soud and Stuehr [40] demonstrated that in the absence of calcium, NOS-1 isolated in the form with oxidized heme (Fe3+) and flavins can be converted into partially reduced form by NADPH; in this form, when the heme remains oxidized both flavins are reduced. Such enzyme binds arginine and slowly (13.6 nmoles/min per mg) oxidizes NADPH by oxygen, but the heme remains oxidized and arginine is not converted, although the enzyme can reduce certain artificial substrates. Addition of Ca2+ results in heme reduction (Fe2+) and dramatic (up to 906 nmoles/min per mg) increase in NADPH oxidation rate. In the presence of arginine, the enzyme generates NO and citrulline, and in the absence of arginine the product is hydrogen peroxide [51]. NADPH oxidation rate at optimal cofactor concentrations is higher than that in the absence of arginine (1450 nmoles/min per mg).
Each NADPH molecule gives two electrons and reduction of the heme iron requires only one electron; thus, the reaction of Abu-Soud--Stuehr (reduction of NOS heme by completely reduced flavins) requires two hemes unless there are no other reducing agents in the system. This process has not been studied in detail; it is not known whether both hemes are reduced simultaneously in a dimeric molecule or the reaction is intermolecular and whether it requires some additional electron transporter (for example, BH4) or the hemes interact directly with each other. Even though the heme is not reduced by free BH4 (potential electron transporter between the hemes) in the absence of calmodulin, this does not indicate that it is not required; the process can be synchronous and BH4 (or another transporter) can influence the oxidation potential after heme reduction if it is bound to the heme.
Calcium--calmodulin enables electron transfer from flavins to the heme and can be considered as a "trigger" of synthetic activity of NOS. Calmodulin binding induces conformational changes in the NOS molecule (increased fluorescence of tryptophane residues and flavins), and this binding influences at least two stages of the electron transfer chain from NADPH to the heme. First, the rate of flavin reduction by NADPH is increased (according to a 20-fold increase in the rate of cytochrome c reduction by the reductase domain of NOS with or without heme); second, the rate of heme reduction by the reductase fragment is increased (see review [71]). NOS-2 is active in the absence of calcium. It was demonstrated [72] that apart from the conserved calcium--calmodulin-binding sequence, there is an additional sequence providing for calmodulin binding at [Ca2+] = 0.
An alternative role for calmodulin, apart from being the trigger coupling two regions of the electron transfer chain, can be increasing the potential of the reductase domain. In this case, low NOS activity in reduction reactions (of heme or nonspecific substrates) can be due to the low potentials of flavins in the absence of calmodulin. Coupling of flavin electron transfer systems can be enough to influence the potential of the reductase domain (see the scheme).
If the data of Abu-Soud and Stuehr are considered as a proof of intramolecular interaction of the hemes, the whole dimeric NOS molecule is a huge pi-electron transfer system (4 flavins, 2 hemes, and possibly 1-2 BH4) forming several hundreds (!) (without pterins: 34 × 22 = 324) different oxidation states and the mean difference E between these states is close to 1 mV. Thus, the complex of the functional NOS molecule should include dozens of various molecules; the properties of such complex (for example, absorption spectrum) would be the sum of their properties. Formation of the system should have impact on the reduction potential of its components.
The three-dimensional structure of the NOS-2 oxygenase domain was resolved at 2.1 Å resolution in 1997 [73], but unequivocal evidence of pi-electron system interaction in dimeric NOS molecules are still lacking.
NOS cannot oxidize NHA in the absence of FMN; this can be considered as evidence for significant differences in the active site (oxidizer) at the first versus the second stage and for changes in flavin potential when they are combined into a common electron system. In fact, if both oxidation stages take place on the same heme, it is difficult to explain the regulation of the chemical behavior of the active Fe--O2 complex (whether in one case, Fe=O is formed and in another case, it does not form). This contradiction is resolved by assuming that BH4 contributes to oxygen activation during the first reaction stage, but this hypothesis evidently is in contradiction with the generally accepted scheme of NOS functioning. In several publications, Austrian scientists demonstrated that BH4 binding by the dimeric NOS molecule is anti-cooperative [6, 74, 75]; however, the consequences of this finding were not compared with the preceding models. In a recent work [75], very interesting results describing a possible influence of BH4 binding on NADPH-oxidase activity of NOS were presented. It looks like NOS belongs to the enzyme class characterized by competitive relationships between all cofactors and substrates.
Unlike the other cofactors, the role of BH4 in NOS is still poorly understood. Arginine and BH4 increase the affinity of each other for the apoenzyme and promote dimerization of the subunits [55]. Moreover, thus formed dimer is exceptionally stable to denaturation due to BH4 [76]. Samples of highly purified enzymes can contain several subfractions differing in the BH4 binding constant.
BH4-deficient NOS synthesizes hydrogen peroxide [51] and its heme is "modified" [77] (i.e., absorption spectrum characteristics that can be associated with modified environment of the iron atom; chemical structure of protoporphyrin was not studied since there is no evidence suggesting its modification). Similar to other BH4-dependent enzymes, the primary structure of NOS has a conservative region responsible for BH4 binding (residues 447-480 in NOS-2). A series of mutant proteins containing single amino acid substitutions in this region was prepared by site-directed mutagenesis. Several mutant enzymes (residues 450 and 453) did not bind BH4 and were inactive [78]. Heterodimers with substitution only in one subunit are active [3]. Thus, only one BH4 molecule is required for formation of active dimeric molecule [74, 75]. If calcium--calmodulin induces the formation of the transfer system, thus changing the potential of the heme--flavin system, paradoxical triggering of NOS activity during Arg and NHA oxidation can be explained by the coupling to BH4 oxidation during the first stage. Indeed, simultaneous oxidation of pterin and arginine would change the length of the coupled system and heme potential and subsequent NHA oxidation might take place without BH4.
Pastor et al. [79] studied the competition of NOS and phenylalanine hydroxylase for BH4. Thus, BH4 participates in competitive interactions of at least two types: it is a competitor by itself and the subject for competition between enzymes.
COMPETITIVE PRODUCTS
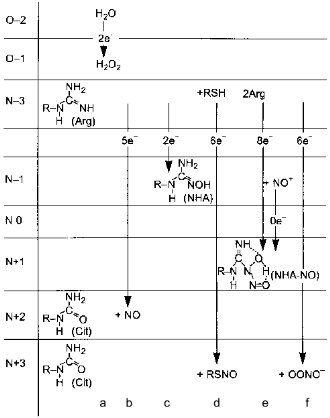
Depending on the concentrations of substrates and cofactors, NOS synthesizes several alternative products (Fig. 8). Their ratios in vivo are unknown and under experimental conditions enzyme assay is usually limited to a single product, i.e., citrulline or NO oxidation products. In the absence of BH4, H2O2 is formed (Bune et al. [80] demonstrated that inhibitors of BH4 synthesis significantly lowers the formation of NO in vivo); deficiency in flavins results in NHA accumulation and arginine deficiency results in appearance of peroxynitrite. However, what does NOS generate when there is plenty of substrates and cofactors is not definitively determined. The problem is due to the inhibition of NOS by its product, NO.
Several alternatives have been discussed: NO can leave the NOS active site as itself, i.e., unmodified; it dissociates as S-nitrosothiol (thionitrite) possibly forming an intermediate with pterin; the product is the NHA--NO complex or peroxynitrite.Fig. 8. Alternative products of NOS. a) In the absence of arginine or tetrahydrobiopterin, NOS oxidizes water forming H2O2. Five-electron oxidation of arginine to NO and citrulline (Cit) (b) and two-electron oxidation forming N-hydroxyarginine (NHA) (c) are competitive and are regulated by flavin availability. Competitive reactions of 6-electron oxidation (formally to NO+) give S-nitrosothiols (RSNO) (d), nitrite (NO2-) (e), or peroxynitrite (OONO-) (f) (under arginine deficiency required to bind superoxide anion that is generated by heme during oxygen reduction; peroxynitrite non-enzymically isomerizes to nitrate). Apparently, NHA is also nitrosylated to N-hydroxy-N-nitrosoarginine (NHA--NO) in an 8-electron process requiring two arginine molecules.
In one of the first publications [81], glutathione was considered as a NOS cofactor. A glutathione-binding region was found in the primary structure of the enzyme. Then, SH-containing compounds were not studied although they were present in the reaction medium as the standard reagents to preserve the enzyme in the native form. In 1995, two independent studies on the role of thiols in NOS-1 activation were published. Japanese scientists [22] worked with rat cerebellum enzyme and German investigators [82] studied the effect of glutathione on porcine brain NOS-1 in detail.
In [22], the enzyme was purified in the absence of SH-reagents. Its activity was low (up to 10 nmoles citrulline/min per mg). Addition of any thiol induced 4-7-fold activation. At concentrations exceeding 2 mM, L-cysteine and glutathione decreased the activity since they began to compete with arginine. (D-Cysteine does not compete with arginine). Addition of dihydropterin reductase enhanced the activity similar to addition of thiols. The enzyme is relatively stable in the absence of thiols, substrate, and cofactors but is rapidly inactivated when addition of the substrates and cofactors is not followed by addition of thiols. At low BH4 concentrations, thiols significantly enhanced the activity; however, even at high BH4 in the absence of thiols, the activity remained low.
Hofmann and Schmidt [82] demonstrated that addition of glutathione enhances citrulline generation by NOS-1, blocks time-dependent inactivation of the enzyme, has some allosteric effect, and influences BH4 consumption (S0.5 was determined). Glutathione had no significant effect on Km for arginine but the values obtained by the authors (7.1-7.4 µM) are slightly higher than the minimal ones reported in the preceding studies (Table 1). The effect on kinetics (but not on enzyme stability) of ascorbate and dihydropterin reductase was similar to that of glutathione. Oxidized glutathione (added to glutathione to change its redox potential) had no effect on arginine oxidation; on the contrary, thioldisulfide isomerase significantly enhanced NOS activity; according to the authors, this suggests that glutathione is important for reduction of the enzyme SH-groups. The effects of other thiols are less notable. NOS was also absorbed on glutathione agarose. Apparently there is no glutathione-binding site on the enzyme since octylglutathione did not compete with glutathione. (Another interpretation can be suggested; for example, a large hydrophobic tail coupled to glutathione molecule can significantly affect the conformation of the peptide (and its aqueous environment) so that it is no longer recognized by the active site; enzyme affinity for octylglutathione can be due to hydrophobic interactions similar to sorption on reverse-phase chromatography matrix).
The interaction of thiols with NOS that does not contain BH4 was studied by Gorren et al. [83]. It was demonstrated that low-molecular-weight thiols bind to the heme (KD = 0, 16, ..., 41 µM) thus enhancing stability and resulting in an absorption spectrum characteristic for bis-thiolate complex (according to spectroscopy, BH4-deficient enzyme in the absence of exogenous thiols is not thiolate). Dithiothreitol binding to the enzyme containing 1 mole BH4 per dimer was a two-stage process (competition of BH4 versus thiol) modulated by L-arginine and thus enzyme-catalyzed generation of citrulline was inhibited. Hence, arginine binds only to the BH4-containing subunit (competition of BH4 versus arginine), whereas the BH4-free subunit does not directly participate in catalysis. On the basis of these data, one cannot exclude a possibility that the heme moiety of the BH4-free subunit influences the redox potential of the system and apparently affects the rapid (not rate-limiting) steps of the whole reaction. A possible role of the other SH-group of dithiothreitol is unknown. Evidently, intermolecular (within a single dimeric NOS molecule) reactivity of the second SH-group can significantly differ from that of the independent thiol molecule in vivo. It would be interesting to study the effects of the length of the carbon chain between SH-groups of dithiols.
Once the NO molecule is synthesized in the NOS active site, it is bound to the heme iron complex; this results in reversible inactivation of the enzyme. Thiols block autoinhibition of NOS and interact with NO forming thionitrites (S-nitrosothiols) upon catalysis by transition metal ions. A possible explanation for thiol requirement for normal functioning of NOS can be their participation in the release of the product, NO, from the active site. In this case, the enzyme should be considered as S-nitrosothiol synthase. If S-nitrosothiol is the end product, six electrons participate in one cycle of nitrogen oxidation because the formal oxidation state of nitrogen is +3; to reduce two oxygen molecules (8 electrons) and fit the electron balance, only one NADPH molecule is required if it is the only coupled reducing agent. This is in apparent contradiction with experimental data on consumption of 1.5 moles of NADPH but it is possible to suggest that S-nitrosothiol can be enzymatically reduced back to NO outside of the heme moiety. Considering the influence of dihydropterin reductase on NOS activity, BH4 can be considered as the site of S-nitrosothiol reduction to NO. It should be noted that BH3 and thiyl radicals (RS·) are odd and short-lived but real molecules of biological systems. Their possible role in NOS catalysis has not been discussed. However, a purely chemical problem of suggesting a system without additional metal ions that could "extract" NO from the heme complex without destroying it can include the BH3--RS combination.
In many cases, biochemical effects of S-nitrosothiols and NO are similar; however, a number of exceptions have been described. For example, Salmonella is not sensitive to NO, but peroxynitrite kills it and S-nitrosothiols are reversible cytostatics [84]. A short review on S-nitrosothiols [85] considers all three variants: NO and S-nitrosothiols are equivalent, NO is active and S-nitrosothiol is inactive, and vice versa. (The structure of endothelium-derived relaxing factor (EDRF) is still a matter of controversy; it was suggested to be NO, S-nitrosothiol, a combination of both, or neither NO nor S-nitrosothiol. Arguments for the former three suggestions were discussed in [85]). A significant feature of NO is its reactivity towards cellular metabolites; the "half-life" of S-nitrosoalbumin in vivo is about 40 min [86], and that of NO is about 5 sec [87].
Similar to equilibrium in the thiol--disulfide system, S-nitrosothiols participate in a transnitrosylation reaction:
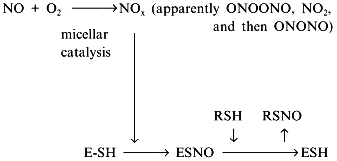
Another contradiction of the NOS mechanisms is enzyme stability towards the effects of NO oxidation products. Oxygen (substrate) and NO (product) are non-polar and are concentrated inside hydrophobic globular protein molecules; the oxidation reaction is a third-order reaction characterized by extremely efficient micellar catalysis [89]. The oxidation products (NOx) formed inside the enzyme molecule are potent nitrosylating agents; hence, the enzyme SH-groups should be converted to S-nitrosothiols. One of the possible functions of thiols in NOS is transnitrosylation resulting in NO released in the form of RSNO with a low-molecular-weight thiol associated with reduction of the SH-groups of the enzyme [90, 91]:
It is accepted that S-nitrosothiols are the main form transporting NO "for long distances". Cysteine, glutathione, and serum albumin have been considered as potential donors of SH-groups. Evidently, several consecutive carriers can participate in the transport of a single NO molecule due to transnitrosylation. Pietraforte et al. [92] studied the role of thiols in NO transport to erythrocytes and critically discussed preceding work (for example, data on formation of S-nitrosothiols and thiyl radicals RS· during their decomposition). Recently, Japanese scientists demonstrated that under aerobic conditions, the half-life of thionitrite of glutathione (GSNO) is decreased by several orders of magnitude by ascorbic acid (seconds versus 10 h in its absence) [93]. Excess glutathione decelerates the decomposition. The concentration of ascorbate in plasma is less than 100 µM and in the cell, it is about 10 mM; hence, plasma half-life of GSNO is high, whereas inside cells it rapidly decomposes.
If S-nitrosothiols are the main form of biogenic NO transformation in the presence of oxidizers and act as the biochemical equivalent of NO, then what is the fraction of S-nitrosothiols in the total pool? (Evidently, depending on the assay, not only NO by itself but its metabolites with similar effect are registered as "NO".) General biochemical considerations suggest that this fraction should be close to unity. This explains significant deviations in estimated concentration of "NO" in vivo. According to Stamler et al. [94], plasma NO and S-nitrosothiol concentrations are about 3 nM and 1 µM, respectively, but erythrocytes play the main role in transporting NO by the blood; a dynamic cycle was suggested involving S-nitrosylation of hemoglobin in the lung and subsequent gradual release of NO to regulate blood pressure [95, 96].
Hecker et al. described the NHA--NO complex that more closely resembled EDRF than NO and S-nitrosothiols do [13, 97]. The chemical structure of the NHA--NO adduct is unidentified. It can be chromatographed, but all attempts to obtain its derivatives resulted in decomposition. According to [13], NHA--NO is formed with high yield in the reaction of stoichiometric amounts of HNO2 with NHA. Thus, if the product is NO derivative (nitrogen oxidation state +2), initial NHA should be oxidized in the reaction. Oxidation of NOH-group nitrogen seems reasonable (see the data of Korth [98] on intermediate formation of iminoxyl radical during NHA oxidation in the NOS active site):
COMPETITION RESULTING FROM HETEROGENEITY OF THE MEDIUM
Earlier studies have suggested one of the strongest arguments against the universal role of biogenic NO in signal transduction, i.e., a kinetic paradox since its high diffusion coefficient and good solubility in aqueous and lipid phases should result in long distance distribution of NO from the source. Chemical reactions can interfere with this distribution. Estimation of mean lifespan of NO in vivo gives values close to 5-30 sec. (NO decomposition in vivo is not a first-order reaction so the term “half-life” is not justified and often results in misunderstanding because "half-life" is intuitively associated with exponential decay; thus, it cannot be used to determine the migration distance (or communication radius) (Gordin and Nedospasov, manuscript in preparation).) At high mean lifespan within the communication zone of a single NO-synthesizing cell, thousands and even millions of adjacent cells [99] would be involved. Discovery of NO equivalents apparently solved the paradox but in fact, the number of contradictions even increased because the rates of NO oxidation and thionitrite formation in vivo and in model reactions in vitro differ by more than one order of magnitude.
Low attention was paid to the impact of the high solubility of NO in hydrophobic phases on kinetics of NO-dependent processes because of the low fraction of these phases in the total volume. However, a unique combination of physicochemical properties of NO dramatically enhances the efficiency of micellar catalysis (kapp/k) of its oxidation by oxygen and of coupled reactions [89]. The lipid phase acts as an NO sponge absorbing NO from the aqueous phase and concentrating it in the small volume of the lipid phase. The effect of the hydrophobic phase fraction of the total volume on the (kapp/k) value has resonance-like maximum at low fractions (about 1%) of the lipid phase close to the in vivo values; thus, the reactions can be accelerated by several hundred fold (Fig. 9). For example, in a two-phase system:
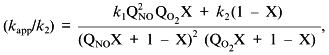
where k1 and k2 are the rate constants in the lipid phase; QNO and QO2 are the partition coefficients of NO and O2 between the phases; X is the volume fraction of the lipid phase. At k1 = k2, QNO = 50, QO2 = 20, and X = 0.01, (kapp/k2) = [(502·20·0.01) + 0.99]/(0.50 + 0.99)2(0.20 + 0.99) ~ (500)/(2.25.1.2) ~ 185.
Thus, at physiological NO concentrations oxidation by oxygen is apparently "slow" but in heterogenous medium in vivo (and low fractions of lipid phase!), it is indeed rapid; on the contrary, rates of reactions in the aqueous phase decrease because NO concentration in this phase is lowered.Fig. 9. Micellar catalysis of NO oxidation by oxygen in a third-order reaction [89]. Maximal acceleration of the reaction (H) corresponds to low (about 1%) fractions of hydrophobic phase (X). The H values rapidly increase at increasing distribution coefficients of the reagents between the phases (Q) (in the plot, the Q values for NO and O2 are equal). The curved line corresponds to the point with maximal H at a given Q. Small changes in X around the maximum result in dramatic differences in the reaction rate.
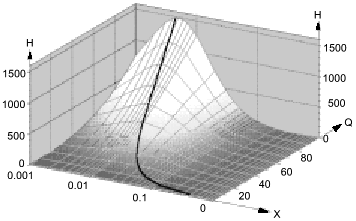
The efficiency of micellar catalysis and the "inhibitory" effect of the NO sponge in the aqueous phase are functions of the partition coefficients (Q) of the reagents between the phases. The Q values depend on electrolyte concentration in the aqueous phase (Nature began to use salting out fractionation long before the chemists started doing it) and on hydrophobicity of the lipid phase; hence, salt and lipid metabolisms are tightly associated with regulation of NO metabolism.
Vanin et al. [100] studied nitrosylation of thiols in aqueous phase catalyzed by iron salts and decomposition of the generated thionitrites. The data can be hardly transferred to in vivo conditions because it is difficult to evaluate the influence of iron complex formation with thiols and other metabolites as well as S-nitrosothiols.
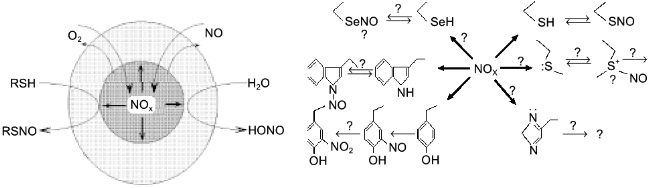
A significant part of biogenic NO is oxidized in hydrophobic phases and the intermediate oxidation products are similar to that of thionitrite formation; lipid membranes, micelles, and hydrophobic protein globules are not only the sites of NO oxidation but the sites of generation of a nitrosylating intermediate in the cell. Hence, proteins are nitrosylated inside, i.e., the phase border acts as a semiconductor filter; it transfers reagents (NO and oxygen) inside the hydrophobic protein globule but does not release the product (NOx) to the outside; the local concentration of NOx inside the protein can be several orders of magnitude higher versus the aqueous environment and the SH-groups of the cysteine residues are thus "intermolecularly" S-nitrosylated in the first turn despite the common excess of thiols (glutathione) in the aqueous phase (Fig. 10).
Apart from cysteine, other amino acid residues can also be nitrosylated. It was unequivocally demonstrated that nitrosylation of tryptophane is reversible; N-nitrosotryptophane (possibly formed via intermediate C-3-nitroso derivative) can act as EDRF if the tryptophane residue is included into plasma proteins [101]. The reduction mechanism is unclear. Under similar conditions (experiments with dipeptides in aqueous solutions), methionine, histidine, and side chains of other amino acids do not react (possibly rapidly decomposed during the experiment?) and selenocysteine has not been studied. On the contrary, nitrosotyrosine is formed but is irreversibly oxidized to nitrotyrosine. Considering significant differences in hydrophobicity and the impact of micellar catalysis on NOx generation, apparent the reactivity of amino acid residues of native proteins should be significantly shifted to the more hydrophobic ones. Thus, phase competition for NO should correspond to competition in protein nitrosylation. So, nitrosylation of protein tryptophane residues is especially intriguing; being the most hydrophobic, they should be located inside the globule and easily nitrosylated. The generated nitrosotryptophane should be stable in the environment of other non-polar residues suggesting that there is an autocatalytic mechanism of NO+ group transfer from the inside of the globule to the aqueous phase. In principle, such catalysis could be performed by the functional groups of residues located adjacent to tryptophan (for example, histidine, cysteine, methionine), but this suggestion requires that the structure of proteins of NO-producing organisms should contain such residue adjacent to tryptophan if the protein preserves its activity in the presence of NO.Fig. 10. On the left, micellar catalysis of NO oxidation. Reagents (NO, oxygen) are concentrated in a hydrophobic protein globule or in a lipid micelle (circle in the center). Generated NOx preferentially nitrosylates groups inside the globule. Reagents react in the aqueous phase only at the phase border; thus, hydrophobic targets are nitrosylated more rapidly among the competitive ones. On the right, nitrosylated amino acid residues. Symbol "?" designates hypothetical and uninvestigated substances and processes.
Mikoyan et al. studied the binding of NO by hydrophilic and hydrophobic iron complexes in vivo [102]. The latter complex was more efficient as expected considering NO lipophilicity.
NO is rapidly oxidized by hemoglobin--O2 and other d-metal complexes forming nitrate and being irreversibly inactivated. Competitive oxidation of NO by oxygen in hydrophobic phases generates nitrite or its derivatives (nitrogen oxidation state +3) which can be reduced back to NO. Thus, heterogeneity of the medium regulates not only nitrosylation/denitrosylation but total NO pool considering the parameters of micellar catalysis.
COMPETITIVE TARGETS
Guanylate cyclase (GC) was historically the first and the most studied target of NO [103]. The heme of the non-activated enzyme has high-spin 5-coordinate iron bound to imidazole of a histidine residue. NO binding (KD = 0.25 µM) converts iron via an intermediate 6-coordinate state to a new 5-coordinate state where iron is bound to NO and histidine is released and can participate in catalysis; this results in 400-fold stimulation of activity. In the case of CO, the reaction is terminated at the octahedric complex stage and imidazole remains bound (for details see [104, 105] and references therein).
The fact that adjacent cells transmit different signal with the help of NO is apparently paradoxical but indeed, this happens rather often. For example, NO simultaneously induces LHRH secretion and ... its shutdown [65, 66] (Fig. 7). The signal from NO-ergic norepinephrine-activated neuron is transmitted to LHRH-neuron and is targeted towards at least two independent targets (ovulation is important and double checking is required): guanylate cyclase and cyclooxygenase (COG); guanylate cyclase increases Ca2+ ion concentration required to activate phospholipase A2 that generates arachidonic acid (AA) from membrane phospholipids; COG converts AA into prostaglandin E2 that activates adenylate cyclase that synthesizes cAMP that induces LHRH secretion. This machinery is triggered by NO as well. An adjacent neuron synthesizing gamma-aminobutyric acid (GABA) is activated (via reaction of NO with GC) at the same time and shortly after that, newly synthesized GABA binds to the specific receptor of LHRH-neuron and blocks the secretion. (For the appreciators of Bacchus: apart from GABA receptors, LHRH-neurons also have ethanol receptors, i.e., ethanol can block transmission of the triggering signal from the NO-neuron [106], so that either ethanol or ovulation but not both simultaneously. Designed perfectly but unfortunately does not always work.)
An interesting hypothesis of NO function was suggested by Brown [107]. Since NO inhibits cytochrome oxidase by competing with oxygen for the active site, increasing NO concentration should influence cellular respiration and levels of ATP and products of its conversion. Hence, various self-regulating systems sensitive to oxygen concentration can be considered (several examples were discussed and explained from this point of view). Finally, cytotoxicity of macrophages and similar NO-producing cells is perfectly explained by blocking cellular respiration.
At the same oxygen concentration depending on NO level, cytochrome oxidase activities in various cells are different as if the enzyme has different Km values for oxygen (see above on competitive substrates). NO degradation also depends on oxygen concentration (for example, oxidation into nitrate) and thus oscillatory systems can be modelled similar to those considered in case of arginine. Their characteristic reaction rates suggest that they should have significantly higher frequency.
Several groups have independently shown that NO induces apoptosis (programmed cell death) [108, 109] but the detected DNA fragmentation did not result from NO or its metabolite directly but depended on protein kinase activity. Recently, Richter et al. [110] studied the link between apoptosis and cellular ATP level, thus supporting the hypothesis of Brown [107].
Association between the competitive mechanisms of NO cytotoxicity (necrosis--apoptosis) and competitive processes of NO oxidation
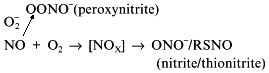
is the subject of extensive studies by the group of Brune [111-116]. The main data were obtained using a series of NO donors.
It is extremely difficult to transfer the data obtained with NO donors to the real situation in vivo. Apart from difficulties in estimation of distribution of NO donors between tissues and their subcellular localization, one should consider their reactions competitive with decomposition and subsequent NO generation as well as the reactions of the intermediate products of this decomposition. Thus, even though S-nitrosothiols can be reduced to NO in vivo, this does not mean that the effects of exogenous S-nitrosothiols are identical to the effects of NO generated during their reduction.
Several competitive processes involving glutathione are discussed in [112], and competition of superoxide anion conversion products is reviewed in [113].
NO (odd) does not react with thiols under standard conditions in the absence of an additional odd molecule or metal ion (numerous publications on such reactions are rarely argued by the authors, but experimental protocols usually contain the description of the source of the third component); however, formation of S-nitrosothiols is one of the main properties of NO. It is believed that protein SH-groups converted to S-nitrosothiols are targets of NO. Possible candidates for the second odd molecule are transition metal ions and another NO molecule. Participation of triplet (usually, "twice odd") O2 is not excluded but this is not enough since even though it is biradical, it is even. Conclusions on the function of NOS in such processes are true even in case of experimental errors, for example, if S-nitrosothiol is the NOS product and modification of the target molecule involves only transnitrosylation (see above about micellar catalysis of S-nitrosothiol synthesis).
Caselli et al. [117] demonstrated that NO inactivates phosphotyrosine phosphatase interacting with an essential SH-group of the enzyme (most experiments were performed using NO donors; the chemistry of the process is not discussed, but other similar studies are reviewed). Inactivation is reversible because addition of thiols reactivates the enzyme. Considering completely anaerobic conditions, it should be assumed that inside the cell (and in most studies of individual enzymes), there is always a one-electron reducer (possibly, transition metal ion complex) which can generate S-nitrosothiol in the presence of excessive SH-compounds. More likely seems that indeed, the conditions were not completely anaerobic (otherwise, how could NO be formed if O2 is a substrate of NOS?) and non-enzymic oxidation of NO by oxygen with the help of micellar catalysis generates NOx. If a protein cysteine residue acts as an SH-group, such modification can result not only in inhibition but activation of the enzyme as demonstrated in the case of p21ras protooncogene (S-nitrosothiol formation involves cysteine and causes conformational changes and binding of GTP instead of GDP [118]). Numerous examples are reviewed in [85].
According to Mayer et al. [119], peroxynitrite non-enzymically converts thiols to S-nitrosothiols with low (1%) yield (apparently, this is catalyzed by metal ions) and guanylate cyclase can be activated by peroxynitrite in the presence of glutathione. Thus, another pathway of S-nitrosothiol generation explains the resistance of NOS to inhibition by NO; it includes formation of the complex of NO with NOS heme, interaction of this complex with superoxide anion (generated by another heme or BH4 of the same NOS molecule or by other means) generating peroxynitrite and reaction of peroxynitrite with thiol.
Nakayama et al. have shown that angiotensin II inhibits NOS induction by cytokines mediated by protein kinase C [120] and Griscavage et al. [121] demonstrated that activation of macrophage NOS biosynthesis is suppressed by protease inhibitors. Thus, the main regulatory pathways involving low-molecular-weight chemical messengers are tightly associated.
If ions and complexes of transition metals are the primary targets of NO, NO can participate in regulation of activity of any biopolymer forming such complexes including metal-dependent enzymes. The existence of numerous forms of ions (differing in distribution of low-molecular-weight ions) implies that there are many NO-dependent enzymes (for example, enzymes known as Zn- or Mg-dependent but functioning in the presence of Mn; this has been studied in pyrophosphatase and RNA-polymerase (Nedospasov, unpublished observations)). Examples of interaction of NO and its conversion products with Fe-S clusters and other metal protein are reviewed in [122]. Surprisingly, only a few systems have been studied.
Exceptional interest of biochemists to NO in recent years has resulted in discoveries of numerous NO-regulated processes whose detailed mechanisms are unknown. For example, it was shown that NO participates in regulation of glucose-dependent metabolism of arachidonic acid [123], possibly via activation of Ca2+-independent phospholipase A2 and prostaglandin synthesis (Fig. 7; for practical application see review of Wink et al. [124], preceding paper [125] and references therein). Peunova and Enikolopov demonstrated the role of NO in cell differentiation [126]. In isolated rat hepatocytes, low doses of NO induce an unknown mechanism defending the cells from subsequent effects of high concentrations of NO [127]. Several hypotheses of heme conversion and intracellular iron metabolism are reviewed in [128, 129]. Malyshev et al. demonstrated that heat shock is associated with enhanced synthesis of NO. NO can participate in induction of heat shock protein (Hsp70) synthesis and these mechanisms may be responsible for regulation of NOS induction [130]. (Feedback on transcription level is discussed by Malyshev and Manukhina [131] and Taylor et al. [4] in this issue.) Lander et al. studied differential activation of phosphokinases by NOx [132]. It was demonstrated that JNK is two orders of magnitude more sensitive than p38 and ERK. They also differ in activation rate, but detailed chemical mechanisms are unknown.
Thus, studies of competition and regulation of activity of NO targets are only beginning and unexpected discoveries are awaiting. Fortunately, the active area is so huge that there should be no competition between the scientists.
REFERENCES
1.Stuehr, D. J., Kwon, N. S., Nathan, C. F.,
Griffith, O. W., Feldman, P. L., and Wiseman, J. (1991) J. Biol.
Chem., 266, 6259-6263.
2.Kwon, N. S., Nathan, C. F., Gilker, C., Griffith,
O. W., Matthews, D. E., and Stuehr, D. J. (1990) J. Biol. Chem.,
265, 13442-13445.
3.Xie, Q.-W., Leung, M., Fuortes, M., Sassa, S., and
Nathan, C. (1996) Proc. Natl. Acad. Sci. USA, 93,
4891-4896.
4.Taylor, B. S., Alarcon, L. H., and Billiar, T. R.
(1998) Biochemistry (Moscow), 63, 766-781.
5.Gorren, A. C. F., and Mayer, B. (1998)
Biochemistry (Moscow), 63, 734-743.
6.Ghosh, D. K., Wu, C., Pitters, E., Moloney, M.,
Werner, E. R., Mayer, B., and Stuehr, D. J. (1997) Biochemistry,
36, 10609-10619.
7.Fukuto, J. M. (1995) Adv. Pharmacol.,
34, 1-15.
8.McCleverty, J. A. (1979) Chem. Rev.,
79, 53-76.
9.Lancaster, J. R., Jr. (1994) Proc. Natl. Acad.
Sci. USA, 91, 8137-8141.
10.Yu, Y., Terada, K., Nagasaki, A., Takiguchi, M.,
and Mori, M. (1995) J. Biochem., 117, 952-957.
11.Hattori, Y., Campbell, E. B., and Gross, S. S.
(1994) J. Biol. Chem., 269, 9405-9408.
12.Nussler, A. K., Billiar, T. R., Liu, Z. Z., and
Morris, S. M., Jr. (1994) J. Biol. Chem., 269,
1257-1261.
13.Hecker, M., Boese, M., Schini-Kerth, V. B.,
Mulsch, A., and Busse, R. (1995) Proc. Natl. Acad. Sci. USA,
92, 4671-4675.
14.Mitchell, J. A., Hecker, M., and Vane, J. R.
(1990) Eur. J. Pharmacol., 176, 253-254.
15.Wu, G., and Brosnan, J. T. (1992) Biochem.
J., 281, 45-48.
16.Nagasaki, A., Gotoh, T., Takeda, M., Yu, Y.,
Takiguchi, M., Matsuzaki, H., Takatsuki, K., and Mori, M. (1996) J.
Biol. Chem., 271, 2658-2662.
17.Reczkowski, R. S., and Ash, D. E. (1994) Arch.
Biochem. Biophys., 312, 31-37.
18.Daghigh, F., Fukuto, J. M., and Ash, D. E. (1994)
Biochem. Biophys. Res. Commun., 202, 174-180.
19.Schmidt, H. H. H. W., Pollock, J. S., Nakane, M.,
Gorsky, L. D., Forstermann, U., and Murad, F. (1991) Proc. Natl.
Acad. Sci. USA, 88, 365-369.
20.Bredt, D. S., and Snyder, S. H. (1990) Proc.
Natl. Acad. Sci. USA, 87, 682-685.
21.Matsuoka, A., Stuehr, D. J., Olson, J. S., Clark,
P., and Ikeda-Saito, M. (1994) J. Biol. Chem., 269,
20335-20339.
22.Komori, Y., Hyun, J., Chiang, K., and Fukuto, J.
M. (1995) J. Biochem., 117, 923-927.
23.Knowles, R. G., Palacios, M., Palmer, R. M. J.,
and Moncada, S. (1989) Proc. Natl. Acad. Sci. USA, 86,
5159-5162.
24.Klatt, P., Schmidt, K., Brunner, F., and Mayer,
B. (1994) J. Biol. Chem., 269, 1674-1680.
25.Furfine, E. S., Harmon, M. F., Paith, J. E., and
Garvey, E. P. (1993) Biochemistry, 32, 8512-8517.
26.Forstermann, U., Pollock, J. S., Schmidt, H. H.
H. W., Heller, M., and Murad, F. (1991) Proc. Natl. Acad. Sci.
USA, 88, 1788-1792.
27.Wolff, D. J., and Gribin, B. J. (1994) Arch.
Biochem. Biophys., 311, 300-306.
28.Olken, N. M., and Marletta, M. A. (1993)
Biochemistry, 32, 9677-9685.
29.Wolff, D. J., and Gribin, B. J. (1994) Arch.
Biochem. Biophys., 311, 293-299.
30.Hevel, J. M., White, K. A., and Marletta, M. A.
(1991) J. Biol. Chem., 266, 22789-22791.
31.Yui, Y., Hattori, R., Kosuga, K., Eizawa, H.,
Hiki, K., and Kawai, C. (1991) J. Biol. Chem., 266,
12544-12547.
32.Evans, T., Carpenter, A., and Cohen, J. (1992)
Proc. Natl. Acad. Sci. USA, 89, 5361-5365.
33.Pollock, J. S., Forstermann, U., Mitchell, J. A.,
Warner, T. D., Schmidt, H. H. H. W., Nakane, M., and Murad, F. (1991)
Proc. Natl. Acad. Sci. USA, 88, 10480-10484.
34.Sessa, W. S., Harrison, J. K., Barber, C. M.,
Zeng, D., Durieux, M. E., D'Angelo, D. D., Lynch, K. R., and Peach, K.
J. (1992) J. Biol. Chem., 267, 15274-15276.
35.Wolff, D. J., and Lubeskie, A. (1995) Arch.
Biochem. Biophys., 316, 290-301.
36.Klatt, P., Schmidt, K., Uray, G., and Mayer, B.
(1993) J. Biol. Chem., 268, 14781-14787.
37.Schmidt, H. H. H. W., and Murad, F. (1991)
Biochem. Biophys. Res. Commun., 181, 1372-1377.
38.Stuehr, D. J., Cho, H. D., Kwon, N. S., Weise,
M., and Nathan, C. F. (1991) Proc. Natl. Acad. Sci. USA,
88, 7773-7777.
39.Kwon, N. S., Nathan, C., and Stuehr, D. (1989)
J. Biol. Chem., 264, 20496-20501.
40.Abu-Soud, H. M., and Stuehr, D. J. (1993)
Proc. Natl. Acad. Sci. USA, 90, 10769-10772.
41.Mayer, B., John, M., Heinzel, B., Werner, E. R.,
Wachter, H., Schultz, G., and Bohme, E., (1991) FEBS Lett.,
288, 187-191.
42.Wang, W. W., Jenkinson, C. P., Griscavage, J. M.,
Kern, R. M., Arabolos, N. S., Byrns, R. E., Cederbaum, S. D., and
Ignarro, L. J. (1995) Biochem. Biophys. Res. Commun.,
210, 1009-1016.
43.Snyder, S. H. (1995) Nature, 377,
196-197.
44.Dinerman, J. L., Dawson, T. M., Schell, M. J.,
Snowman, A., and Snyder, S. H. (1994) Proc. Natl. Acad. Sci.
USA, 91, 4214-4218.
45.Huang, P. L., Dawson, T. M., Bredt, D. S.,
Snyder, S. H., and Fishman, M. C. (1993) Cell, 75,
1273-1286.
46.Huang, P. L., Huang, Z., Mashimo, H., Bloch, K.
D., Moskowitz, M. A., Bevan, J. A., and Fishman, M. C. (1995)
Nature, 377, 239-242.
47.Bune, A. J., Shergill, J. K., Cammack, R., and
Cook, H. T. (1995) FEBS Lett., 366, 127-130.
48.Lantoine, F., Brunet, A., Bedioui, F., Devynck,
J., and Devynck, M.-A. (1995) Biochem. Biophys. Res. Commun.,
215, 842-848.
49.Chenais, B., Yapo, A., Lepoivre, M., and Tenu,
J.-P. (1993) Biochem. Biophys. Res. Commun., 196,
1558-1565.
50.Sennequier, N., and Stuehr, D. J. (1996)
Biochemistry, 35, 5883-5892.
51.Heinzel, B., John, M., Klatt, P., Bohme, E., and
Mayer, B. (1992) Biochem. J., 281, 627-630.
52.Pou, S., Pou, W. S., Bredt, D. S., Snyder, S. H.,
and Rosen, G. M. (1992) J. Biol. Chem., 267,
24173-24176.
53.Baggiolini, M., and Wyman, M. P. (1990) Trends
Biochem. Sci., 15, 69-72.
54. Nedospasov, A. A., Lifanov, A. P., and Rodina,
E. V. (1994) Biochemistry (Moscow), 59, 1119-1125.
55.Abu-Soud, H. M., Loftus, M., and Stuehr, D. J.
(1995) Biochemistry, 34, 11167-11175.
56.Mayer, B., Klatt, P., Werner, E. R., and Schmidt,
K. (1995) J. Biol. Chem., 270, 655-659.
57.Mayer, B., and Werner, E. R. (1995) Naunyn
Schmiedebergs Arch. Pharmacol., 351, 453-463.
58.Mayer, B., and Werner, E. R. (1995) Adv.
Pharmacol., 34, 251-261.
59.Afework, M., Tomlinson, A., Belai, A., and
Burnstock, G. (1992) Neuroreport, 3, 893-896.
60.Brenner, C., and Fuller, R. S. (1992) Proc.
Natl. Acad. Sci. USA, 89, 922-926.
61.Steiner, D. F., Smeekens, S. P., Ohagi, S., and
Chan, S. J. (1992) J. Biol. Chem., 267, 23435-23438.
62.Lindberg, I. (1991) Mol. Endocrinol.,
5, 1361-1365.
63.Vernigora, A. N., and Gendin, M. T. (1995)
Biochemistry (Moscow), 60, 1953-1963 (Russ.).
64.Dang, C. V., Glinski, R. L., Gainay, P. C., and
Hilderman, R. H. (1982) Biochemistry, 21, 1959-1966.
65.Rettori, V., Belova, N., Dees, W. L., Nyberg, C.
L., Gimeno, M., and McCann, S. M. (1993) Proc. Natl. Acad. Sci.
USA, 90, 10130-10134.
66.Seilicovich, A., Duvilanski, B. H., Pisera, D.,
Theas, S., Gimeno, M., Rettori, V., and McCann, S. M. (1995) Proc.
Natl. Acad. Sci. USA, 92, 3421-3424.
67.Gelperin, A. (1994) Nature, 369,
61-63.
68.Venema, R. C., Ju, H., Zou, R., Ryan, J. W., and
Venema, V. J. (1997) J. Biol. Chem., 272, 1276-1282.
69.Stuehr, D. J., and Ikedo-Saito, M. (1992) J.
Biol. Chem., 267, 20547-20550.
70.Klatt, P., Pfeiffer, S., List, B. M., Lehner, D.,
Glatter, O., Bachinger, H. P., Werner, E. R., Schmidt, K., and Mayer,
B. (1996) J. Biol. Chem., 271, 7336-7342.
71.Stuehr, D. J., Abu-Soud, H. M., Rousseau, D. L.,
Feldman, P. L., and Wang, J. (1995) Adv. Pharmacol., 34,
207-213.
72.Ruan, J., Xie, Q. W., Hutchinson, N., Cho, H.,
Wolfe, G. C., and Nathan, C. (1996) J. Biol. Chem., 271,
22679-22686.
73.Crane, B. R., Arvai, A. S., Gachhui, R., Wu, C.,
Ghosh, D. K., Getzoff, E. D., Stuehr, D. H., and Tainer, J. A. (1997)
Science, 278, 425-431.
74.Gorren, A. C., Schrammel, A., Schmidt, K., and
Mayer, B. (1997) Biochemistry, 36, 4360-4366.
75.List, B. M., Klosch, B., Volker, C., Gorren, A.
C., Sessa, W. C., Werner, E. R., Kukovetz, W. R., Schmidt, K., and
Mayer, B. (1997) Biochem. J., 323, 159-165.
76.Klatt, P., Schmidt, K., Lehner, D., Glatter, O.,
Bachinger, H. P., and Mayer, B. (1995) EMBO J., 14,
3687-3695.
77.Wang, J., Stuehr, D. J., and Rousseau, D. L.
(1995) Biochemistry, 34, 7080-7087.
78.Cho, H. J., Martin, E., Xie, Q., Sassa, S., and
Nathan, C. (1995) Proc. Natl. Acad. Sci. USA, 92,
11514-11518.
79.Pastor, C. M., Williams, D., Yoneyama, T.,
Hatakeyama, K., Singleton, S., Naylor, E., and Billiar, T. R. (1996)
J. Biol. Chem., 271, 24534-24538.
80.Bune, A. J., Brand, M. P., Heales, S. J. R.,
Shergill, J. K., Cammack, R., and Cook, H. T. (1996) Biochem.
Biophys. Res. Commun., 220, 13-19.
81.Stuehr, D. S., Kwon, N. S., Cho, H. J., and
Nathan, C. F. (1990) Biochem. Biophys. Res. Commun., 168,
558-565.
82.Hofmann, H., and Schmidt, H. H. H. W. (1995)
Biochemistry, 34, 13443-13452.
83.Gorren, A. C., List, B. M., Schrammel, A.,
Pitters, E., Hemmens, B., Werner, E. R., Schmidt, K., and Mayer, B.
(1996) Biochemistry, 35, 16735-1674545.
84.De Groote, M. A., Granger, D., Xu, Y., Campbell,
G., and Prince, R. (1995) Proc. Natl. Acad. Sci. USA, 92,
6399-6403.
85.Upchurch, G. R., Jr., Welch, G. N., and Loscalzo,
J. (1995) Adv. Pharmacol., 34, 343-349.
86.Stamler, J. S., Simon, D. I., Osborne, J. A.,
Mullins, M. E., Jaraki, O., Michel, T., Singel, D. J., and Loscalzo, J.
(1992) Proc. Natl. Acad. Sci. USA, 89, 444-448.
87.Lowenstein, C. J., and Snyder, S. H. (1992)
Cell, 70, 705-707.
88.Dicks, A. P., Swift, H. R., Williams, D. L. H.,
Butler, A. R., Al-Sadoni, H. H., and Brian, G. C. (1996) J. Chem.
Soc., Perkin Trans. II, 741-745.
89.Gordin, V. A., and Nedospasov, A. A. (1998)
FEBS Lett., 424, 239-242.
90.Wink, D. A., Nims, R. W., Darbyshire, J. F.,
Christodoulou, D., Hanbauer, I., Cox, G. W., Laval, F., Laval, J.,
Cook, J. A., Krishna, M. C., DeGraff, W. G., and Mitchell, J. B.
(1994) Chem. Res. Toxicol., 7, 519-525.
91.Goldstein, S., and Czapski, G. (1996) J. Am.
Chem. Soc., 118, 3419-3425.
92.Pietraforte, D., Mallozzi, C., Scorza, G., and
Minetti, M. (1995) Biochemistry, 34, 7177-7185.
93.Kashiba-Iwatsuki, M., Yamaguchi, M., and Inoue,
M. (1996) FEBS Lett., 389, 149-152.
94.Stamler, J. S., Jaraki, O., Osborne, J. A.,
Simon, D. I., Keaney, J., Vita, J. A., Singel, D., Valeri, C. R., and
Loscalzo, J. (1992) Proc. Natl. Acad. Sci. USA, 89,
8087-8091.
95.Jia, L., Bonaventura, C., Bonaventura, J., and
Stamler, J. S. (1996) Nature, 380, 221-226.
96.Gow, A. J., and Stamler, J. S. (1998)
Nature, 391, 169-173.
97.Hecker, M., Sessa, W. C., Harris, H. J., Anggard,
E. E., and Vane, J. R. (1990) Proc. Natl. Acad. Sci. USA,
87, 8612-8616.
98.Korth, H.-G., Sustmann, R., Thater, C., Butler,
A. R., and Ingold, K. U. (1994) J. Biol. Chem., 269,
17776-17779.
99.Wood, J., and Gartwaite, J. (1994)
Neuropharmacology, 33, 1235-1244.
100.Vanin, A. F., Malenkova, I. V., and
Serezhenkov, V. A. (1997) Nitric Oxide: Biol. Chem., 1,
191-203.
101.Zhang, Y. Y., Xu, A. M., Nomen, M., Walsh, M.,
Keaney, J. F., and Loscalzo, J. (1996) J. Biol. Chem.,
271, 14271-14279.
102.Mikoyan, V. D., Kubrina, L. N., Serezhenkov, V.
A., Stukan, R. A., and Vanin, A. F. (1997) Biochim. Biophys.
Acta, 1336, 225-234.
103.McDonald, L. J., and Murad, F. (1995) Adv.
Pharmacol., 34, 263-275.
104.Stone, J. R., and Marletta, M. A. (1995)
Biochemistry, 34, 14668-14674.
105.Deinum, G., Stone, J. R., Babcock, G. T., and
Marletta, M. A. (1996) Biochemistry, 35, 1093-1099.
106.Santeros, G., Rettori, V., Franchi, A., Genaro,
A., Cebrae, E., Faletti, A., Gimeno, M., and McCann, S. M. (1995)
Proc. Natl. Acad. Sci. USA, 92, 3416-3420.
107.Brown, G. C. (1995) FEBS Lett.,
369, 136-139.
108.Shimaoka, M., Iida, T., Ohara, A., Taenaka, N.,
Mashimo, T., Honda, T., and Yoshiya, I. (1995) Biochem. Biophys.
Res. Commun., 209, 519-526.
109.Messmer, U. K., Lapetina, E. G., and Brune, B.
(1995) Mol. Pharmacol., 47, 757-765.
110.Richter, C., Schweizer, M., Cossarizza, A., and
Franceschi, C. (1996) FEBS Lett., 378, 107-110.
111.Haendeler, J., Messmer, U. K., Brune, B.,
Neugebauer, E., and Dimmeler, S. (1996) Shock, 6,
405-409.
112.Sandau, K., and Brune, B. (1996) Cell.
Signal., 8, 173-177.
113.Sandau, K., Pfeilschifter, J., and Brune, B.
(1997) J. Immunol., 158, 4938-4946.
114.Messmer, U. K., Reed, U. K., and Brune, B.
(1996) J. Biol. Chem., 271, 20192-20197.
115.Messmer, U. K., and Brune, B. (1996) Arch.
Biochem. Biophys., 327, 1-10.
116.Brune, B., Sandau, K., and Von Knethen, A.
(1998) Biochemistry (Moscow), 63, 817-825.
117.Caselli, A., Camici, G., Manao, G., Moneti, G.,
Pazzagli, L., Cappugi, G., and Ramponi, G. (1994) J. Biol.
Chem., 269, 24878-24882.
118.Lander, H. M., Ogiste, J. S., Pearce, S. F. A.,
Levi, R., and Novogrodsky, A. (1995) J. Biol. Chem., 270,
7017-7020.
119.Mayer, B., Schrammel, A., Klatt, P., Koesling,
D., and Schmidt, K. (1995) J. Biol. Chem., 270,
17355-17360.
120.Nakayama, I., Kawahara, Y., Tsuda, T., Okuda,
M., and Yokoyama, M. (1994) J. Biol. Chem., 269,
11628-11633.
121.Griscavage, J. M., Wilk, S., and Ignarro, L. J.
(1995) Biochem. Biophys. Res. Commun., 215, 721-729.
122.Drapier, J.-C., and Bouton, C. (1996)
BioEssays, 18, 549-556.
123.Gross, R. W., Rudolph, A. E., Wang, J.,
Sommers, C. D., and Wolf, M. J. (1995) J. Biol. Chem.,
270, 14855-14858.
124.Wink, D. A., Vodovotz, Y., Cook, J. A.,
Krishna, M. C., Kim, S., Coffin, D., DeGraff, W., Deluca, A. M.,
Liebmann, J., and Mitchell, J. B. (1998) Biochemistry (Moscow),
63, 802-809.
125.Kelner, M., and Uglik, S. F. (1994) Arch.
Biochem. Biophys., 312, 240-243.
126.Peunova, N., and Enikolopov, G. (1995)
Nature, 375, 68-73.
127.Kim, Y. M., Bergonia, H. A., and Lancaster, J.
R. (1995) FEBS Lett., 374, 228-232.
128.Kim, Y. M., Bergonia, H., Muller, C., Pitt, B.
R., Watkins, W. D., and Lancaster, J. R. (1995) Adv. Pharmacol.,
34, 277-291.
129.Pantopoulos, K., Weiss, G., and Hentze, M. W.
(1994) Trends Cell Biol., 4, 82-86.
130.Malyshev, I. Yu., Malugin, A. V., Golubeva, L.
Yu., Zenina, T. A., Manukhina, E. B., Mikoyan, V. D., and Vanin, A. F.
(1996) FEBS Lett., 391, 21-23.
131.Malyshev, I. Yu., and Manukhina, E. B. (1998)
Biochemistry (Moscow), 63, 840-853.
132.Lander, H. M., Jacovina, A. T., Davis, R. J.,
and Tauras, J. M. (1996) J. Biol. Chem., 271,
19705-19709.