REVIEW: Inducible Nitric Oxide Synthase in the Liver: Regulation and Function
B. S. Taylor*, L. H. Alarcon, and T. R. Billiar
Department of Surgery, University of Pittsburgh, Pittsburgh, PA 15261, USA; fax: 412-648-1172; E-mail: BSTA@med.pitt.edu* To whom correspondence should be addressed.
Received September 1, 1997
The inducible nitric oxide synthase (iNOS) gene is expressed by hepatocytes in a number of physiologic and pathophysiologic conditions affecting the liver including septic and hemorrhagic shock. The molecular regulation of iNOS expression is complex and occurs at multiple levels in the gene expression pathway. The cytokines TNF-alpha, IL-1beta, and INF-gamma synergistically activate iNOS expression in the liver, and the human iNOS gene was first cloned from cytokine-stimulated hepatocytes. iNOS expression requires the transcription factor NF-kappaB and is down-regulated by steroids, TGF-beta, the heat shock response, p53, and nitric oxide (NO) itself. In vivo, hepatic iNOS induction is differentially regulated from the typical acute-phase reactants and is not expressed as a mandatory component of the acute phase response. Thus, numerous mechanisms have evolved to regulate iNOS expression during hepatocellular injury. Studies of the effects of NO in the liver demonstrate that induced NO synthesis plays an important role in hepatocyte function and protects the liver during sepsis and ischemia reperfusion. Its cytoprotective role is best exemplified in a rodent model of endotoxemia. Here the addition of the nonspecific NOS inhibitors significantly increased hepatic damage. NO exerts a protective effect through its ability to prevent intravascular thrombosis by inhibiting platelet adhesion and neutralizing toxic oxygen radicals. NO also exerts a protective effects both in vivo and in vitro by blocking TNF-alpha-induced apoptosis and hepatotoxicity, in part by a thiol-dependent inhibition of caspase-3-like protease activity. These studies demonstrate the cytoprotective effects of NO in the liver and suggest hepatic iNOS expression functions as an adaptive response to minimize inflammatory injury. In addition, NO has anti-tumor effects as well as known mutagenic effects, is involved in the systemic vasodilatation of cirrhosis, and has potent antimicrobial properties.
KEY WORDS: nitric oxide, inducible nitric oxide synthase, gene regulation, hepatocytes, apoptosis, ischemia reperfusion, malaria, cirrhosis, cancer
Abbreviations: NO) nitric oxide; NOS) nitric oxide synthase; NADPH) reduced nicotinamide adenine dinucleotide phosphate; O2) molecular oxygen; FAD) flavin adenine dinucleotide; FMN) flavin mononucleotide; BH4) tetrahydrobiopterin; NHA) NG-hydroxy-L-arginine; NO2-) nitrite; NO3-) nitrate; cNOS) constitutive nitric oxide synthase; iNOS) inducible nitric oxide synthase; ncNOS) neuronal cell nitric oxide synthase; ecNOS) endothelial cell nitric oxide synthase; mRNA) messenger ribonucleic acid; NF-kappaB) nuclear factor-kappaB; TNF-alpha) tumor necrosis factor-alpha; IL-1) interleukin-1; IFN-gamma) interferon-gamma; LPS) lipopolysaccharide; TGF-beta) transforming growth factor-beta; L-NAME) NG-nitro-L-arginine-methyl ester; L-NMMA) NG-monomethyl-L-arginine; EDRF) endothelium-derived relaxing factor; cGMP) cyclic guanosine monophosphate; O2-) superoxide anion; N2O3) dinitrogen trioxide; NO+) nitrosonium ion; OONO-) peroxynitrite; DNA) deoxyribonucleic acid; OH·) hydroxyl radical; SNAP) S-nitroso-N-acetyl-penicillamine; cAMP) cyclic adenosinemonophosphate; SNP) sodium nitroprusside; L-NIL) N-iminoethyl-L-lysine; Hsp70) heat shock protein 70; GSH) glutathione; L-NNA) NG-nitro-L-arginine; HBV) hepatitis B virus; SVR) systemic vascular resistance.
Nitric oxide (NO), the end product of the enzyme nitric oxide synthase
(NOS), is a potent biologic mediator whose diverse physiologic and
pathophysiologic properties have been recognized over the last decade.
NO has a broad range of biological activities and plays a role in
regulating blood flow, neurotransmission, antimicrobial defense
mechanisms, and in immunomodulation [1]. The
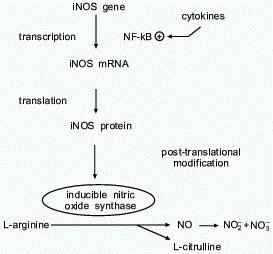
L-arginine-NO biosynthetic pathway was first identified in 1987 [2-4] and is shown in Fig. 1. Once produced, NO has a short half life
(t1/2 = seconds) and undergoes spontaneous oxidation
to the inactive metabolites nitrite and nitrate
(NO2- and NO3-). This
chapter reviews the regulation and function of the inducible NO
synthase (iNOS) isoform in the liver. The regulation of iNOS is complex
and occurs at multiple levels in the pathway of gene expression
including signal transduction, transcription, translation, and via
post-translational modifications and cofactor requirements. Also, the
basic biology and physiology of NO will be reviewed, and the potential
roles of NO in regulating hepatic function and disease will be
discussed.
Fig. 1. NO biosynthetic pathway. Inducible nitric oxide synthase catalyzes the oxidation-reduction reaction of L-arginine, in the presence of molecular oxygen, to form the free radical NO and citrulline. NO has a short half-life and is oxidized to the inactive end-products nitrite and nitrate (NO2- and NO3-). iNOS expression is upregulated by cytokines through the transcription factor NF-kappaB. Transcriptional and post-transcriptional mechanisms are important in iNOS regulation.
MOLECULAR REGULATION OF NITRIC OXIDE SYNTHASE IN THE LIVER
The liver plays a central role in a large number of metabolic and immune processes. It has become clear in recent years that normal and pathologic liver functions depend on the complex interactions among the heterogenous sub-populations of liver cells. The liver is composed of the true hepatic parenchymal cells (hepatocytes), vascular endothelial cells, resident hepatic macrophages (Kupffer cells), bile duct cells, and fat-storing cells (Stellate or Ito cells). Each of these cell types control an array of physiologic responses and influence neighboring cells in a number of ways. Kupffer cells belong to the monocyte lineage and account for 80-90% of the tissue macrophages in the body. Lining the liver sinusoids, they are positioned near the endothelial cells and maintain proximity to the hepatocytes via the peri-sinusoidal space of Disse [5]. While anatomically the liver is composed of structural units centered around the hepatic vascular supply, from a physiologic standpoint it is organized in functional units consisting of parenchymal cells and their adjacent Kupffer cells, which are engaged in a variety of cell--cell interactions. As induced NO production has been documented in all of these cell types under various conditions, it stands to reason that NO would be involved in many of these metabolic processes.
Rodent macrophages were among the first cell types that were found capable of producing NO from iNOS. In response to LPS with or without IFN-gamma, iNOS mRNA and protein are produced, and large amounts of NO2- and NO3- are detected [6]. Rat Kupffer cells are similar to other macrophages in their ability to respond to inflammatory stimuli with iNOS induction [7]. It was later demonstrated that hepatocytes produce NO [8]; this was the first evidence that a parenchymal cell type could express iNOS. Hepatocytes readily express iNOS in response to conditioned medium from LPS and IFN-gamma-stimulated Kupffer cells [8]. It has been shown that the Kupffer cell secretory products responsible for the induction of iNOS in hepatocytes are TNF-alpha and IL-1beta [9], and it appears that IL-1beta is the more important stimulus [10]. These Kupffer cell-derived cytokines act synergistically with LPS and IFN-gamma to substantially induce iNOS expression in hepatocytes [9, 11]. In LPS-primed hepatocytes [12], or rats injected with turpentine to produce remote tissue injury [13], single cytokines such as TNF-alpha, IL-1 or even LPS alone are sufficient to induce NO synthesis. In experimental models of in vivo endotoxemia, hepatocytes readily express iNOS mRNA, which can be detected by Northern analysis within 3 h, and peak in iNOS enzyme levels at 12-16 h. Plasma NO2- and NO3- subsequently decline rapidly [14].
Hepatocytes express large amounts of iNOS in response to killed Corynebacterium parvum (C. parvum) injection, a known inducer of various cytokines such as IL-1, TNF-alpha, and IFN-gamma in combination with LPS [14]. Other stimuli that induce expression of iNOS are prostaglandins [15] and protein kinase C activators such as phorbol esters and the Ca2+ ionophore A23187 [12]. Dexamethasone and hydrocortisone prevent the LPS-induced expression of iNOS in hepatocytes [16]. Other factors that suppress iNOS expression in hepatocytes include the NO donor S-nitroso-N-acetyl-DL-penicillamine (SNAP) [17], hepatocyte mitogens hepatocyte growth factor (HGF), epidermal growth factor (EGF) and TGF-beta [18].
Hepatic endothelial cells [19] and Ito cells [20] can also generate NO through iNOS expression. Furthermore, like in the rodent liver, human hepatocytes produce iNOS in response to a number of inflammatory cytokines [21]. (The first human iNOS gene was cloned from primary human hepatocytes by our group [22].) Thus, hepatocytes can be exposed to NO derived from neighboring Kupffer, endothelial and Ito cells as well as autogenously-derived NO.
Compared to other organs which express iNOS, the liver is unique in that it contains a complete urea cycle and an enzyme, phenylalanine hydroxylase, which compete for the NOS cofactor tetrahydrobiopterin (BH4). The urea cycle synthesizes arginine from ammonia and ornithine. Although this arginine is rapidly hydrolyzed to urea and ornithine by arginase, it was unknown whether “leakage” of arginine from the urea cycle represented a source of substrate for hepatocyte iNOS. A study by Wettstein and coworkers [23] using isolated perfused livers from rats pretreated with LPS exhibited a measurable increase in NO release when the perfusate buffer was supplemented with ammonium chloride to increase flux through the urea cycle. In contrast, perfused livers from either endotoxin- or C. parvum-treated rats perfused with glutamine as a nitrogen donor did not demonstrate any increase in NO2- and NO3- release, despite a large increase in ureagenesis [24]. Interestingly, there was ongoing NO production in the absence of added arginine. Thus, an endogenous source of arginine exists in hepatocytes, but leakage of arginine from the urea cycle makes a relatively small contribution.
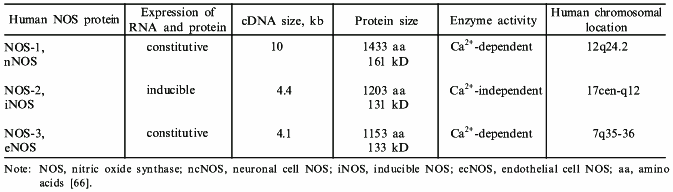
The inducible NOS (iNOS) cDNA has been cloned from mouse [25-27], rat [28, 29], and human cells [22, 30-32]. Our group isolated the first human iNOS cDNA from LPS- and cytokine-stimulated primary human hepatocytes [22]. The sequence of the human hepatocyte iNOS clone reveals a 4145 base pair cDNA containing a 3459 base pair open reading frame which encodes a polypeptide of 1153 amino acids with a calculated molecular mass of 131 kD. The ATG start site at nucleotide 207 conforms to the critical requirements of the Kozak consensus sequence [33]. Compared to the murine macrophage iNOS, the hepatocyte iNOS sequence is six amino acids longer in the amino portion of the protein and three amino acids longer at the carboxyl terminus. Similar to other NOS isoforms, hepatocyte iNOS contains consensus recognition sites for the cofactors FMN, FAD, and NADPH in the carboxyl half of the protein which have been shown to be important for iNOS enzyme activity. In addition, a consensus sequence for a calmodulin recognition site is also present. Overall, human iNOS displays a 80% sequence identity to murine macrophage iNOS at both the nucleotide and amino acid levels. Compared to human endothelial cNOS [34] and human neuronal cNOS [35], human hepatocyte iNOS displays 51 and 53% amino acid sequence identity, respectively. Functional analysis of the human iNOS cDNA was performed by insertion of the human iNOS cDNA into the pCIS expression vector (Genetech) and following transient transfection in human kidney cells, substantial NOS activity was detected by conversion of radiolabelled arginine to citrulline [22]. The calcium chelating agent EDTA and the calmodulin antagonist trifluoperazine decreased NOS activity by 50-65%. These results were surprising since cNOS enzymes are calcium/calmodulin dependent, while rodent iNOS contains a tightly bound calmodulin which is not calcium sensitive [36]. It is likely that low levels of calcium are required for optimal calmodulin binding to human iNOS.
Once the human iNOS cDNA clone was isolated, the probe was used to directly study the induction of iNOS mRNA in human hepatocytes. Northern blot analysis was performed on RNA from cultured human hepatocytes stimulated with LPS, TNF-alpha, IL-1beta, and IFN-gamma for 2-48 h. No iNOS mRNA was detected in the unstimulated human hepatocytes [21]. However, a single mRNA band at ~4.5 kb first appeared 4 h after cytokine and LPS stimulation, peaked at 8 h, and was barely detectable by 48 h. To correlate mRNA levels with iNOS activity, NO2- and NO3- levels were measured from the culture supernatants and were found to increase 20- to 30-fold at 24 and 48 h after stimulation.
After the initial report of iNOS expression in primary human hepatocytes [21], a variety of human cells and cell lines were shown to express iNOS (Table 1). Subsequently, human iNOS cDNAs were cloned from chondrocytes [31], DLD-1 colon carcinoma cell line [32], and A-172 glioblastoma cell line [33]. In each case, these cDNAs were found to be identical (>99% sequence homology) to the human hepatocyte iNOS cDNA. While most cell types require a combination of cytokines to activate iNOS expression, IL-1beta alone at high doses can induce iNOS mRNA in cultures of primary human hepatocytes [10] and chondrocytes [30]. In general, however, the amount of NO produced by human cells is lower than that seen with their rodent counterparts.
Table 1. Cell and tissue types that express constitutive and inducible nitric oxide synthases
| Constitutive NOS (NOS-1 or -3) | Inducible NOS (NOS-2) |
|---|---|
| Adrenal gland | Chondrocytes |
| Endocardium | Endocardium |
| Glial cells | Eosinophils |
| Mast cells | Fibroblasts |
| Megakaryocytes | Glial cells |
| Mesangial cells | Hepatocytes |
| Myocardium | Keratinocytes |
| Neurons | Kupffer cells |
| Peripheral nerves | Lung epithelial cells |
| Platelets | Lymphocytes |
| Respiratory epithelium | Macrophages/monocytes |
| Vascular endothelial cells | Megakaryocytes |
| Mesangial cells | |
| Myocardial cells | |
| Neutrophils | |
| Osteoblasts | |
| Pancreatic islet cells | |
| Placental trophoblasts | |
| Retinal epithelial cells | |
| Vascular endothelial cells | |
| Vascular smooth muscle cells | |
| Cell lines | |
| A-172 glioblastoma cell | |
| A549 lung epithelial cell | |
| AKN-1 liver epithelial cell | |
| B-cell lymphoma line | |
| CaCO-2 adenocarcinoma line | |
| DLD-1 adenocarcinoma line | |
| MG 63 osteosarcoma line | |
| SW480/SW620 colon cancer line |
Next, in collaboration with J. S. Mudgett, at Merck Research Laboratories, the human iNOS gene was isolated. To determine the size and structure of the human iNOS gene, a human fibroblast genomic library was screened using iNOS cDNA [37]. To summarize, two rounds of screening yielded seven distinct cosmid clones which were aligned by restriction mapping, Southern blot hybridization, and DNA sequencing analysis. All of the isolated clones were found to be part of a single genomic locus. The full-length human iNOS gene is 37 kb in length and is composed of 26 exons and 25 introns (Table 2). This genomic structure is similar to that of human endothelial and neuronal NOS genes, and suggests the divergence from a common “ancestral” gene. Primer extension analysis of LPS and cytokine-stimulated human hepatocyte RNA identified the transcriptional initiation site 30 base pairs downstream of the TATA box. By utilizing a somatic cell hybrid mapping panel and fluorescent in situ hybridization, the human iNOS gene was mapped to chromosome 17 at position 17 cen-q11.2 [37]. The human neuronal and endothelial NOS reside on chromosome 12 and 7, respectively, confirming that the three NOS genes are distinct.
Table 2. Nitric oxide synthase isoforms:
designations and characteristics
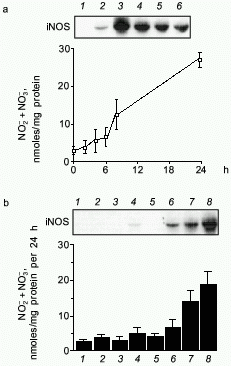
To study the transcriptional regulation of the human iNOS gene, we then sought to characterize the human iNOS promoter [38]. To facilitate this work, a human liver epithelial cell line (derived from histologically normal human liver and designated AKN-1) was isolated. Immunohistochemical staining and karyotype analysis revealed a transformed liver cell line with both biliary and epithelial cell features. When stimulated with the cytokine mix (CM) of TNF-alpha, IL-1beta, and INF-gamma, iNOS mRNA was observed at 2 h, peaked at 4 h, and diminished by 24 h (Fig. 2). A time dependent rise in NO release was seen that lagged behind the iNOS mRNA expression. Of the individual cytokines tested, only IFN-gamma weakly stimulated AKN-1 mRNA expression. Double cytokine combinations had additive or synergistic effects, and the combination of all three cytokines induced the highest levels of iNOS mRNA. These results are similar to the regulation of the iNOS gene in primary human hepatocytes, indicating that the AKN-1 cell line was likely to be useful for studying the regulation of human iNOS gene expression. We then tested the hypothesis that the human iNOS gene is regulated in part by gene transcription. Nuclear run-on analysis in the AKN-1 cells showed a fivefold increase in cytokine induced transcriptional activity of iNOS (Fig. 3) [38]. This up-regulation appears to be mediated by the transactivating transcription factor NF-kappaB in both human (unpublished data) and rat hepatocytes [17, 42].
Fig. 2. Cytokine-induced iNOS expression in AKN-1 cells. a) Time course of iNOS mRNA induction (Northern blot, upper) (control (1), cytokine mix 2 (2), 4 (3), 6 (4), 8 (5), and 24 h (6)) and nitrite and nitrate (NO2- and NO3-) production in culture supernatants (lower) following stimulation with TNF-alpha (1000 u/ml) + IL-1beta (100 u/ml) + IFN-gamma (250 u/ml). b) Pattern of 6-h iNOS mRNA induction (Northern blot, upper) (control (1), TNF-alpha (2), IL-1beta (3), IFN-gamma (4), TNF-alpha and IL-1beta (5), TNF-alpha and IFN-gamma (6), IL-1beta and IFN-gamma (7), and cytokine mix (8)) and NO2- and NO3- release (lower) in response to cytokines and cytokine combinations. Values for NO2- and NO3- are expressed as mean ± SEM for three independent experiments ([38], permission granted).
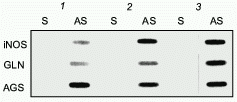
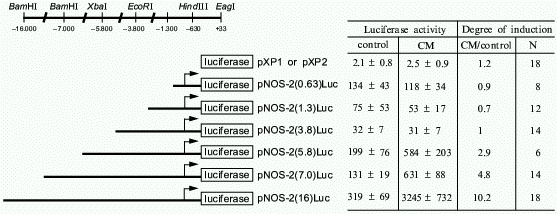
To delineate the cytokine-responsive regions of the human iNOS promoter, ~16 kb of the human iNOS gene 5´ flanking region was isolated and deletional iNOS-luciferase promoter constructs were generated. Transient transfections into AKN-1 cells revealed that the first 3.8 kb upstream demonstrated basal promoter activity, but failed to show any cytokine-inducible activity. However, 3- to 5-fold increases in promoter activity were seen in constructs extending up to -5.8 and -7.0 kb, and a cytokine-mediated 10-fold increase in promoter activity was seen after transfection of a -16 kb construct (Fig. 4). To confirm that regions upstream from -3.8 kb contain cytokine-inducible elements whose function are independent of elements other than the basal iNOS promoter, heterologous thymidine kinase promoter constructs were generated from -3.8 to -5.8, -5.8 to -7.0, and -7.0 to -16.0 kb. Stimulation of these heterologous constructs with CM after transfection into AKN-1 cells increased the thymidine kinase activity 2-, 2-, and 7-fold, respectively [38]. This confirmed the presence of cytokine-responsive regulatory elements upstream from -3.8 kb. These results contrast markedly with those reported for the murine macrophage iNOS promoter [39, 40]. Here, Xie reported that only 1 kb of the proximal 5´ flanking region is required to confer LPS and cytokine inducibility [39], indicating that regulation of the human iNOS gene is unique compared to the murine iNOS promoter.Fig. 3. Nuclear run-on analysis of cytokine-induced iNOS message in AKN-1 cells. Control cells were not exposed to cytokines (1). Cytokine-stimulation (cytokine mix, CM) for 2 (2) and 4 h (3) was conducted with TNF-alpha (1000 u/ml) + IL-1beta (100 u/ml) + IFN-gamma (250 u/ml). Nuclei were isolated and incubated with radiolabelled CTP. 32P-Labelled nuclear RNA was hybridized to a GeneScreen membrane containing 5 µg of immobilized sense (S) and antisense (AS) cRNA probes of iNOS, glutaminase (GLN), and argininosuccinate synthetase (AGS) ([38], permission granted).
Besides defining what factors upregulated iNOS gene expression, our laboratory is interested in defining those factors which augment iNOS expression. Glucocorticoids are known to inhibit induced NO synthesis in several cell types [2]. We found that dexamethasone decreases iNOS mRNA levels in rat hepatocytes and that this down-regulation occurs at the level of transcription [41]. This effect is a result of decreased iNOS transcription due to the ability of dexamethasone to upregulate I-kappaBalpha expression and inhibit cytokine stimulated NF-kappaB activity [42]. Furthermore, while I-kappaBalpha levels are increased by dexamethasone, nuclear p65 levels are decreased. This indicates that dexamethasone may be inhibiting iNOS gene expression by inhibiting NF-kappaB nuclear translocation and through directly binding to nuclear p65. It is interesting that the effect of dexamethasone appears to be tissue and species specific. For example, Kleinert et al. found that dexamethasone inhibited cytokine-induced iNOS mRNA synthesis in human A549 epithelial cells by 70% and that this inhibition was mediated in part by down-regulating activation of NF-kappaB without an increase in I-kappaBalpha mRNA levels [43].Fig. 4. Deletional analysis of the human iNOS gene promoter in cytokine-stimulated human liver cells. The 5´ deletional constructs generated are shown along with the specific restriction sites. AKN-1 cells were stimulated with cytokine mix (CM): TNF-alpha (1000 u/ml) + IL-1beta (100 u/ml) + IFN-gamma (250 u/ml) after transient transfections with the promoter constructs. Luciferase activity is expressed as light u/mg of protein in cell lysates. Values are expressed as mean ± SEM, N is number of experiments ([38], permission granted).
Additional studies have demonstrated that the induction of the heat shock response by hyperthermia or sodium arsenite exposure blocked subsequent iNOS expression in AKN-1 human liver cells [44] as well as in primary rat hepatocytes [45]. The inhibitory effect of heat shock on iNOS expression was observed in promoter transfection experiments, however there was no effect of heat shock response on iNOS enzyme activity in AKN-1 cells transduced to stably express iNOS. These results indicate that prior induction of the heat shock response inhibits iNOS gene transcription, but not iNOS translation or enzyme activity. These findings underscore the complex array of phenotypic responses in the liver during stress conditions. A similar heat shock protein 70 mediated suppression of iNOS expression was reported in brain astroglial cells [46]. We believe that induction of the heat shock response is an adaptive defense mechanism that may prevent over-expression of iNOS during certain inflammatory conditions. This is supported by the observation that high-level NO exposure stimulated heat shock expression [47].
In other studies, the effects of hepatocellular mitogens on cytokine-induced iNOS expression in cultures of primary human hepatocytes were examined. Hepatocyte growth factor (HGF), epidermal growth factor (EGF), and transforming growth factor TGF-alpha all down-regulated LPS and cytokine-induced human iNOS mRNA, iNOS enzyme activity, and NO release [48]. Interestingly, the cytokine-stimulated NO release caused a decrease in hepatocyte protein and DNA synthesis, and this effect was partially reversed by the liver growth factors. These mitogens are known to directly trigger hepatic DNA and protein synthesis; however, our results raise the possibility that these hepatic growth factors may also promote protein and DNA synthesis during inflammatory conditions, in part, by suppressing iNOS expression. The precise mechanism by which these growth factors exert their effects on iNOS expression is unknown.
In collaboration with C. Harris, at the NIH Laboratory of Human Carcinogenesis, the relationship between iNOS expression and the tumor suppressor gene product p53 was investigated [49]. NO has been shown to cause DNA strand breaks as well as mutations in human cells. Since p53 plays an important role in cellular response to DNA damage from exogenous mutagens, the hypothesis that p53 may perform a similar role in regulating iNOS expression was tested. Exposure of human cells to a NO donor resulted in p53 accumulation. Also, expression of p53 in multiple human cell lines, including the AKN-1 liver cell line, resulted in a down-regulation of iNOS gene expression by inhibiting the iNOS promoter. These data imply a negative feedback loop where NO-induced DNA damage activates p53 expression which then mediates repression of the human iNOS gene.
Recently, we have shown that NO inhibits its own gene expression by down-regulating NF-kappaB expression in rat hepatocytes [17] and primary human hepatocytes (unpublished data). In the presence of the NO donor SNAP or via endogenous NO production, NO down-regulates inducible NO levels as well as iNOS mRNA and protein production. The work presented in this study adds to the complexity of iNOS regulation by demonstrating that as levels of NO increase, feedback regulation begins and NO negatively modulates NF-kappaB DNA binding activity and enzyme activity to tightly regulate the amount of NO produced. Hepatocytes can be induced to synthesize large quantities of NO and negative feedback regulation serves to attenuate this physiologic response to possibly prevent further tissue injury. In summary, this data identifies a novel negative feedback loop whereby NO down-regulates iNOS gene expression, possibly to limit over production during pathophysiologic conditions. By understanding the molecular regulation of iNOS expression, therapeutic modalities can be designed to govern its induction. The nuclear transcription factor NF-kappaB is one potential target for such anti-inflammatory therapy.
In addition to transcriptional control, post-transcriptional regulation may also be important in the regulation of iNOS expression in the liver. The 3´-UTR of the human hepatocyte iNOS cDNA contains several AT-rich sequences which correspond to the AU sequences in the mRNA. These ATTTA sequence motifs have been shown in many labile cytokine and proto-oncogene transcripts and have been shown to destabilize mRNA [50]. A related sequence, TTATTTAT, was identified in the 3´-UTR of TNF and other cytokine genes [51] and has also been implicated in destabilizing mRNA as well as inhibiting translational efficiency [52]. Currently, studies are in progress to determine whether these “AU”-rich elements are involved in the regulation of iNOS mRNA stability.
POST-TRANSLATIONAL REGULATION OF iNOS
In addition to the transcriptional and post-transcriptional control of the iNOS genes, significant post-translational regulation of the iNOS protein exists. All three isoforms share features of post-translational modification, and some of these characteristics are summarized in Table 2. Mechanisms of post-translational control include protein stability, dimerization, phosphorylation, subcellular localization, cofactor binding, and availability of the substrates L-arginine and O2.
Experiments have also been conducted to examine the rate-limiting aspect of BH4 availability in perfused livers and isolated hepatocytes [53]. Suppression of BH4 levels markedly reduced the conversion of phenylalanine to tyrosine by phenylalanine hydroxylase but had little impact on iNOS activity. Furthermore, phenylalanine increased BH4 synthesis as previously described [54] while arginine had no effect. These findings are not entirely unexpected because the iNOS Km for BH4 is 100-fold lower than the phenylalanine hydroxylase Km for BH4. Thus, the requirement of iNOS for BH4 is much lower. While phenylalanine hydroxylase relies on allosteric upregulation of BH4 synthesis by the substrate, iNOS does not.
THE EFFECTS OF NITRIC OXIDE ON HEPATOCYTE FUNCTION
A large body of evidence supports the notion that NO is an important regulator of hepatocyte function (Table 3). Various lines of investigation have shown that NO inhibits cultured hepatocyte protein synthesis. When stimulated by inflammatory mediators or glucocorticoids, hepatocytes secrete a class of products called acute phase proteins [55]. These proteins maintain a number of homeostatic functions during the inflammatory process. However, when hepatocytes are cocultured with Kupffer cells and the system is exposed to LPS, a significant reduction in the incorporation of [3H]leucine occurs, signifying an inhibition of hepatocyte protein synthesis [56]. This inhibition of protein synthesis is accompanied by increased levels of NO2- and NO3- in the supernatant, and can be prevented with L-NMMA [57], confirming that NO produced by Kupffer cells and hepatocytes can act to reduce the total protein production of hepatocytes. The addition of exogenous NO or NO donors reproduces the NO-mediated inhibition of protein synthesis [58]. Likewise, when isolated hepatocytes in primary culture are exposed to the appropriate cytokine mix, a similar decrease in protein synthesis occurs, associated with elevation in NO2- and NO3- levels. These changes are also inhibited with L-NMMA, demonstrating that endogenous hepatocyte NO synthesis can act in an autocrine fashion to inhibit protein synthesis in vitro [9]. In the coculture system and in primary hepatocytes in isolated culture, the production of NO appears to be the result of iNOS because there is a delay of several hours between stimulation and the release of NO [59].
Table 3. NO-mediated effects on
hepatocytes
Inhibition of protein synthesis
Inhibition of gluconeogenesis
Inhibition of glycogenolysis
Inhibition of glyceraldehyde-3-phosphate dehydrogenase
Activation of soluble guanylate cyclase
Inhibition of cytochrome P-450 1A1 and 1A2
Inhibition of mitochondrial respiration
Inhibition of aconitase
The mechanism of NO-mediated suppression of hepatocyte protein synthesis remains unknown. It does not involve loss of hepatocyte viability, because it is reversible with NOS inhibitors, the hepatocytes exclude trypan blue, and no increase in hepatocellular enzyme release is detected [58]. cGMP analogs do not reproduce the inhibition of protein synthesis, thus the mechanism appears to be cGMP-independent [58].
It remains unclear whether NO-mediated suppression of hepatocyte protein synthesis occurs in vivo. In a sepsis-induced model of iNOS expression, increased hepatocyte protein synthesis has been demonstrated [60]. Others have used a cecal ligation and puncture model and found a decrease in the level of in vivo hepatic protein synthesis with administration of NOS inhibitors [61]. These results conflict with our in vitro work. Thus, determining the role of NO on hepatic protein synthesis will require further investigation.
The liver is an important organ in the metabolism of carbohydrates. Liver glycogen metabolism is controlled by hormones such as glucagon and epinephrine. Preliminary data from in vitro work suggests that NO mediates an inhibitory effect on carbohydrate metabolism in hepatocytes. Brass and coworkers showed that the NO donor S-nitroso-N-acetyl-penicillamine (SNAP) inhibits cyclic adenosine monophosphate (cAMP) and glucagon-stimulated hepatic glycogenolysis [62], suggesting that glucose homeostasis may be partially controlled by NO. Furthermore, Horton and coworkers demonstrated that NO donors inhibit cultured hepatocyte gluconeogenesis [63] raising the possibility that NO could account for the suppressed gluconeogenesis encountered in models of sepsis. In other in vitro work, we and others have demonstrated a marked NO-dependent inhibition of the activity of glyceraldehyde-3-phosphate dehydrogenase in rat livers with high-level iNOS expression [64]. However, we were unable to demonstrate an NO-dependent suppression of gluconeogenesis in LPS-treated rats using the NOS inhibitor L-NMMA (nonspecific NOS inhibitor) or aminoguanidine (iNOS-specific inhibitor). Similarly, no difference in the LPS-mediated decrease in hepatic gluconeogenesis was seen between iNOS knockout and iNOS wild-type mice, making the role of NO in suppression of gluconeogenesis questionable [65]. Thus, while NO has been shown to inhibit hepatic gluconeogenesis in cell culture, there is to date no evidence for this in vivo. These results underscore the importance of confirming in vitro data with in vivo experiments.
One of the earliest actions of NO to be defined was that NO binds avidly to heme prosthetic groups and the iron sulfur complexes in certain enzymes [66]. This interaction may result in enzyme activation or inhibition. In the liver, a number of enzymes are potential targets of this type of interaction. Like in other cell types, soluble guanylate cyclase activity in hepatocytes is regulated by NO. In response to NO-generating compounds, rat liver slices release cGMP [67], while cultured hepatocytes stimulated to express iNOS produce moderate amounts of cGMP [68]. While all of the roles of cGMP in hepatocyte physiology have not been determined, we have recently shown that NO-stimulated cGMP production inhibits TNF-alpha-induced apoptosis in hepatocytes [69]. High levels of NO inhibit hepatocyte mitochondrial respiration in vitro [70, 71]. However, proof that NO inhibits mitochondrial function in vivo is lacking.
The ability of the liver to metabolize a large number of drugs, toxins, carcinogens, and other metabolic products depends on the activity of the heme-containing cytochrome P-450 class of proteins [72]. Cytochrome P-450 activity in hepatocytes is inhibited in coculture with cytokine-stimulated Kupffer cells which produce NO [73]. Furthermore, inflammatory conditions that induce iNOS in hepatocytes also result in inhibition of cytochrome P-450 activity. The administration of L-NAME prevents the inhibition of cytochrome P-450 in LPS-stimulated rats [74] and in hepatocytes stimulated to produce NO [75]. Thus, both exogenous and endogenous NO can affect the activity of this important enzyme system, and this may explain the clinical observation of altered hepatic metabolism of certain drugs and toxic substances in the setting of systemic inflammatory processes and liver dysfunction.
THE CYTOPROTECTIVE ROLE OF NO IN THE LIVER
In conditions such as the systemic inflammatory response or sepsis a number of profound metabolic changes occur. Endotoxemia and systemic inflammation can result in liver damage as detected histologically and by the presence of liver enzymes in the plasma. In these settings iNOS is upregulated throughout the liver and especially in cells such as hepatocytes and Kupffer cells [14]. The finding of liver iNOS induction in sepsis is also supported in humans. Elevated NO2- and NO3- levels are detected in the plasma of septic patients [76]. A large volume of experimental work has attempted to determine the role of NO in sepsis in the liver and in other organs, namely the vasculature, heart, lung, and kidney. In the field of NO biology, both beneficial and detrimental NO-mediated properties have been described.
In various experimental models, our group has shown that nonspecific inhibition of the NOS enzyme in endotoxemia results in increased liver damage, supporting a beneficial role of NO in the liver during sepsis. In an in vivo murine model of endotoxemia-induced hepatic necrosis, increased NO production was hepatoprotective, while NOS inhibition resulted in markedly increased hepatic injury [77]. In this study, killed C. parvum injection resulted in the development of hepatitis, which was then followed 5-7 days later by the administration of LPS, causing liver necrosis. L-NMMA administration resulted in increased hepatic injury, with a 3- to 5-fold increase in liver damage as detected by ornithine carbamyltransferase (OCT) and aspartate aminotransferase (AST). This data suggests that NO produced locally plays a protective role in the liver during systemic inflammation. Additional studies using the same model of hepatic injury showed that co-administration of superoxide dismutase could attenuate the L-NMMA-associated exacerbation of hepatic injury [78], while cyclooxygenase inhibition worsened the injury [79]. These results suggests that reactive oxygen intermediates play a prominent role in the LPS-induced hepatic cytotoxicity. Also, prostaglandin synthesis appears to act in synergism with NO in its hepatoprotective role in the face of an LPS challenge. Histologic examination of the liver during endotoxemia with or without NOS inhibition shows increased microvascular thrombosis in mice treated with NOS inhibitors [78, 80], confirming the known role of NO in maintaining vascular patency and preventing platelet aggregation and leukocyte adhesion to the endothelium. However, Szabo and coworkers [81] showed that the selective iNOS inhibitors reduced LPS-induced damage in the rat liver. They postulated that while low-level production of NO via cNOS was cytoprotective and maintained microvascular patency, excess NO production via iNOS mediated cytotoxicity.
To further examine the role of the specific NOS isoforms we have carried out experiments where nonselective or iNOS-selective inhibitors were infused continuously into the liver of rats during endotoxemia. We found that the nonselective inhibitors (L-NMMA or L-NAME) increased both necrosis and apoptosis of hepatocytes, while the iNOS-specific inhibitors N-iminoethyl-L-lysine (L-NIL) or aminoguanidine increased only apoptosis during endotoxemia [82]. We have subsequently shown that NO has a potent anti-apoptotic effect in hepatocytes and inhibits hepatocyte apoptosis resulting from nutrient withdrawal [69]. NO appears to exert these actions by both cGMP-dependent and independent mechanisms. Therefore, we find little evidence for direct NO-mediated hepatotoxicity in endotoxemia. It appears that during sepsis, low-level NO production in the liver (via cNOS) protects against hepatocyte necrosis, while higher amounts of NO (produced by iNOS) prevent apoptosis.
The mechanisms of endotoxemia-induced hepatocyte apoptosis are beginning to be understood. In response to stimuli such as the TGF-beta1 [83], Fas ligand [84], or TNF-alpha and D-galactosamine [85], hepatocytes will undergo the characteristic apoptotic changes such as chromosomal condensation, oligosomal DNA fragmentation and cytoplasmic blebbing. While in some cell types NO may induce apoptosis [86, 87], there is a growing body of evidence that NO plays an anti-apoptotic role in the liver. In vitro studies aimed at determining the mechanism of this action have confirmed the previously described in vivo work. Fetal hepatocytes in primary culture (pre-stimulated with LPS to induce iNOS) are protected from TGF-beta1-mediated apoptosis [88]. NO donors are also protected against apoptotic cell death in these cultured cells. Similarly, sodium nitroprusside confers complete protection against TNF-alpha-induced hepatocyte apoptosis in D-galactosamine-sensitized mice in vivo [89]. Several mechanisms for the anti-apoptotic effect of NO have been postulated. Our group has recently shown that the NO donor SNAP induces the expression of heat shock protein 70 (Hsp70) mRNA and protein in hepatocytes, which confers protection from TNF-alpha and actinomycin D-induced apoptosis [47]. The degree of protection correlated directly with the level of Hsp70 expression, and could be reversed by blocking Hsp70 expression with an antisense oligonucleotide to Hsp70. It was also suggested that causing glutathione oxidation was the mechanism by which NO induced Hsp70. Another line of investigation implicates NO in inhibiting the activity of caspase-3-like protease. This pro-apoptotic enzyme family belongs to the protease signaling cascade. Hepatocytes in primary culture were stimulated to undergo apoptosis by either removal of growth factors, exposure to TNF-alpha, or anti-Fas antibody. Inducing iNOS with cytokine mix or in the presence of exogenous NO donors substantially reduced the loss of cell viability and completely eliminated the appearance of DNA fragmentation [90]. This was associated with inhibition of caspase-3-like activity by S-nitrosylation and an indirect suppression of caspase-3-like activation via a cGMP-dependent process.
Kuo and coworkers have provided evidence that NO also protects against acetaminophen-induced toxicity. In this model, inhibition of NO synthesis worsened the resultant liver injury [91]. In this study, L-NMMA administration prior to acetaminophen resulted in a doubling of the plasma aspartate aminotransferase, with a concomitant decrease in cellular glutathione (GSH) levels, suggesting that the inhibition of NO synthesis potentiates acetaminophen hepatotoxicity by depleting GSH stores, perhaps reducing the cellular ability to neutralize oxidant stress.
There is also evidence to suggest that NO may be beneficial in the liver under a number of pathologic conditions where reactive oxygen intermediates are produced. These substances include superoxide anion (O2-), hydroxyl radical (OH·), and hydrogen peroxide (H2O2). In the liver, these reactive species can cause direct membrane and DNA damage, lipid peroxidation, and hepatocyte necrosis [92]. Under these redox conditions, NO may act as an antioxidant by reacting with toxic oxygen metabolites, producing less toxic species [93]. This may help explain the data that show a protective role for NO in murine LPS-induced hepatic necrosis, because reactive oxygen intermediates produced by LPS-stimulated macrophages are known to cause this injury [94]. Furthermore, Kim, Bergonia, and Lancaster have shown that pretreating hepatocytes with NO renders the cells less sensitive to a subsequent exposure to H2O2 or high concentrations of NO [95]. NO-induced heme oxygenase, which may occur through the liberation of iron within the cell, then provides protection through the formation of biliverdin. Thus the induction of the protective mechanisms requires relatively high concentrations of NO. However, under other conditions, NO reacts with these same reactive species to produce toxic intermediates. This is the case with the production of peroxynitrite (OONO-) resulting from the reaction of NO and O2-. Peroxynitrite can oxidize sulfhydryl groups, cause lipid peroxidation, nitration of tyrosines, and DNA base damage, and can decompose to generate other potent oxidants [96]. Thus, the balance of NO protective and cytotoxic effects will depend, in part on the redox state of the cell.
THE EFFECT OF NITRIC OXIDE ON LIVER FUNCTION DURING ISCHEMIA AND
REPERFUSION
Liver ischemia--reperfusion is an important syndrome encountered in a number of clinical scenarios including trauma, hemorrhagic shock, liver resection, and liver transplantation. The generation of reactive oxygen intermediates during the reperfusion phase underlies the pathophysiology of this syndrome. The effects of NO will depend on the context of its production, namely the presence and relative amounts of other reactive intermediates with which NO may interact and the antioxidant status of the tissue. Furthermore it is probably important to differentiate between warm and cold ischemia.
Early evidence for the interaction between NO and O2- in the liver during ischemia--reperfusion was provided by Bautista and Spitzer who showed that inhibition of NO in the isolated perfused liver increased oxygen radical release [97]. However, Ma and coworkers showed that increased expression of iNOS induced by LPS pretreatment markedly increased liver damage in a NO-dependent manner [98]. Using iNOS knockout mice we have found that the absence of iNOS confers protection from liver damage in both hemorrhagic shock and warm ischemia and reperfusion (unpublished results). It is likely that the combination of NO and O2- forms peroxynitrite resulting in toxicity in warm ischemia and reperfusion. Suppression of glutathione levels in this insult probably also contributes to the injury. Cold ischemia and reperfusion may suppress the expression of iNOS or preserve the antioxidant systems and therefore may not exhibit the same NO-dependent injury.
In models of intestinal ischemia--reperfusion, it has been shown NO donors improve survival and decrease intestinal myeloperoxidase activity in the intestine [99], suggesting that NO inhibits neutrophil adherence and migration in the intestinal microcirculation. It was thought that similar effects might be seen in the liver; however, no such benefit was seen in a rodent model of hepatic ischemia--reperfusion [100] or one model of ischemia--reperfusion plus LPS challenge [101].
Hemorrhagic shock represents a common clinical problem faced by clinicians. Decompensated hemorrhagic shock is reached at a volume of blood loss and duration of hypotension after which the vasculature remains hyporesponsive to volume resuscitation or pressors. This syndrome is accompanied by organ dysfunction, including liver injury. In a rodent model of decompensated hemorrhagic shock, infusion of a non-selective inhibitor L-NAME caused an increase in the shock-induced hepatic injury as detected by plasma liver enzymes and liver histology [102]. However, infusion of the iNOS selective inhibitor N-(iminoethyl)-L-lysine reduces hepatic damage (unpublished). Thus, the constitutive NOS is essential to preserve perfusion, where as induced NO contributes to hepatic injury.
More direct evidence for a protective role for constitutive NO in hepatic ischemia--reperfusion was demonstrated in a rat model of occlusion of the left and medial hepatic lobe vessels for 1 h, followed by reperfusion for 1 or 24 h. In this study, rats pretreated with the NOS inhibitor NG-nitro-L-arginine (L-NNA) developed worsening of endothelial cell and hepatocyte injury, increased release of hepatocyte enzymes, increased lipid peroxidation, and reduction in hepatic blood flow. These effects were abolished by pretreatment with L-arginine [103]. Similarly, a two-hit model of hepatic ischemia--reperfusion followed by administration of LPS results in significant activation of Kupffer cells and subsequent liver injury [104]. In this model, administration of L-NAME worsened the hepatic injury and further impaired microvascular flow; this could be reversed with concomitant infusion of L-arginine or a NO donor. These results are contrasted by our work where iNOS inhibitors reduce hepatic damage in warm ischemia--reperfusion. Similar results were obtained when iNOS knockout mice were compared to wild-type mice (unpublished). Thus, similar to hemorrhagic shock, iNOS expression contributes to damage in isolated ischemia--reperfusion, where as constitutive NO is protective.
THE ROLE OF NITRIC OXIDE IN LIVER TUMORS
NO has anti-tumor effects as well as known mutagenic effects. NO and reactive oxygen species in concert with chronic inflammation may initiate or enhance carcinogenesis in humans. Infection by bacteria, parasites, or viruses and tissue inflammation such as gastritis, hepatitis and colitis are recognized risk factors for human cancers. In the liver, both viral and helminth infections have been closely associated with the development of tumors. Ohshima et al. have documented the occurrence of cholangiocarcinoma in the presence of liver fluke infestation [105]. Liu et al., in a woodchuck hepatitis virus model of chronic hepatitis, demonstrated the possibility that viral hepatitis increases the risk of liver cancer through a mechanism of increased NO production [106]. In humans, NO levels are elevated in patients with chronic hepatitis and this has been linked with the predisposition to develop liver cancer [107].
The process of carcinogenesis is thought to be a step-wise process involving the inactivation of tumor suppressor genes and the activation of oncogenes by either mutations or deletions of the DNA. Subsequent to DNA damage, cell division must occur for a tumor to develop. NO and its reactive derivatives may play an active role in the multistage process of carcinogenesis by direct cellular cytotoxicity and mutagenicity. The mechanisms of NO-induced cytotoxicity and mutagenicity are numerous and include nitrosative deamination, DNA strand breakage by NO2-, oxidative deamination by peroxynitrite, and DNA modification by metabolically activated N-nitrosamines. The formation of N2O3 leads to the formation of N-nitroso compounds which deaminate and crosslink DNA. N2O3 can also inactivate important enzymes involved in DNA repair mechanisms. As discussed earlier, NO reacts with superoxide anion to form the cytotoxic radical peroxynitrite which is also capable of decomposing to hydroxyl radicals and nitrogen dioxide. The generation of peroxynitrite and other oxidizing agents has also been shown to be genotoxic [108]. Interestingly, DNA deamination results in specific mutations including GC to AT. Sequence data on gene mutations from HBV isolated from chronically infected patients reveal the same GC to AT mutation. This same mutation is evident in the p53 gene from patients with liver cancer. The DNA repair molecule p53 plays an important role in the cellular response to DNA damage from ionizing radiation, UV light, and both exogenous and endogenous chemical mutagens. Forrester et al. demonstrated that endogenous NO induces DNA damage, and results in p53 accumulation, which through a negative feedback mechanism inhibits iNOS gene expression. This implies that p53 expression is upregulated in the presence of excessive NO production to prevent NO-induced DNA damage [49]. These data suggest that hepatocytes possess a mechanism to prevent the deleterious effects of NO in the development of liver tumors.
In addition to its carcinogenic potential, it has been shown that NO (from activated macrophages) can kill tumor cells [109]. The target for NO-mediated anti-tumor actions are iron and sulfur containing enzymes involved in mitochondrial respiration. However, whether NO acts as a tumor surveillance mechanism in the liver is unclear. Data from Stadler et al. [70], showed that exogenous NO markedly suppressed mitochondrial aconitase as well as complex I and complex II enzymes, two components of the electron transport chain, while endogenously generated NO was less effective. Fukumura and coworkers demonstrated in an in vivo and ex vivo model using a co-culture system with a hepatoma cell line and Kupffer cells, that induced NO from Kupffer cells produced mitochondrial dysfunction followed by cell membrane disruption leading to tumor cell death [110]. These results suggest that the liver, via Kupffer cell interactions, possesses a mechanism of tumor cell recognition and eradication.
NO AND CIRRHOSIS
Portal hypertension is associated with hemodynamic disturbances in the systemic and splanchnic circulations. In the chronic phase, liver cirrhosis progressively results in a dilated splanchnic system and systemic hemodynamic changes. These include an elevated heart rate and cardiac index as well as decreased systemic vascular resistance (SVR). A number of disturbances seen in cirrhosis can be explained by the “underfill theory”. These include the development of ascites, edema, and the hepatorenal syndrome. According to this theory, a decrease in effective plasma volume related to peripheral vasodilation underlies the etiology of these changes. The decreased effective perfusion volume stimulates the volume receptors, leading to activation of the renin-angiotensin and sympathetic systems, and release of atrial natriuretic factor. These changes cause renal sodium retention and edema, and may lead to decreased glomerular filtration and the development of the hepatorenal syndrome [111].
Several mediators of systemic vasodilation in cirrhosis have been proposed [112]. Growing evidence supports the LPS-induced NO production and consequent vasodilation in cirrhosis. Elevated serum levels of endotoxin are detectable in a large number of cirrhotic patients [113, 114], even in the absence of clinical signs of infection. The mechanism is thought to involve the abnormal porto-systemic shunting [115], allowing the bacterial LPS normally cleared by the liver to enter the systemic circulation. This may explain why the hemodynamic effects of cirrhosis are similar to endotoxic sepsis. Elevated NO production in cirrhosis is supported by the finding of increased serum and urine NO3- levels [116] and increased urinary cGMP levels [117] in humans with cirrhosis, with a direct correlation between severity of hemodynamic changes and levels of NO3-. There is also increased levels of cGMP in aortic segments of cirrhotic rats, which is reduced with chronic L-NAME administration [118]. In animals, glucocorticoids prevent the decrease in SVR in cirrhosis presumably by inhibiting NO production [119]. Further evidence to support this theory was obtained in a rat model of chronic portal hypertension induced by portal vein ligation. In this model of cirrhosis, inhibition of NO synthesis causes significant vasoconstriction in both the systemic and splanchnic beds [120], supporting the role of NO in mediating these derangements. This work was confirmed by studies in carbon tetrachloride-induced cirrhosis in rats, where L-NAME administration corrected the systemic hyperdynamic changes [121]. The vascular production of NO, as measured by aortic cGMP production, was correspondingly reduced in the cirrhotic rats treated with L-NAME.
The data in humans, however, is incomplete. Ongoing work will establish the roles of NOS inhibition and anti-endotoxin therapy in the management of patients with portal hypertension and hyperdynamic systemic circulation. It should also be noted that not all of the NO produced in cirrhosis is detrimental. In animal models of ethanol liver toxicity, inhibition of NOS resulted in increased hepatic microvascular vasoconstriction and hepatocellular damage [122].
ANTIMICROBIAL ACTIONS OF NITRIC OXIDE IN THE LIVER
Nitric oxide has been shown to be a potent antimicrobial agent against various bacterial, protozoan, parasitic, viral, and helminthic infections [123]. The utility of NO as a antimicrobial agent was first recognized by the Sumerians, who used nitrite to cure meats. The commercial food industry later exploited its inhibitory effects on Clostridium sporogenesis in the prevention of botulism in canned food products [124]. Since 1987, when Granger et al. [125] showed that L-arginine is essential for the microbiostatic activity of macrophages, it has been shown that NO is associated with extracellular [126-128] and intracellular [123, 129] mediated cytotoxicity. Table 4 identifies the bacterial, parasitic, and viral agents against which NO has antimicrobial activity [130].
Table 4. Pathogens sensitive to NO-mediated
antimicrobial effects
Cryptococcus neoformans
Schistosoma mansoni
Trypanosoma brucei
Trypanosoma cruzi
Toxoplasma gondie
Mycobacterium avium
Leishmania major
Plasmodium vinckei
Plasmodium yoelii
Plasmodium berghei
Plasmodium falciparum
Staphylococcus aureus
Ectromelia virus
Vaccinia virus
Herpes simplex virus type I
Epstein--Barr virus
Note: Reviewed in [130].
The mechanism by which NO exerts its antimicrobial effects are varied and relate to its small size and lipophilicity. The toxicity of NO against pathogens is typically a result of the elevated and sustained production of NO elicited by cytokine stimulation and by bacterial, fungal, protozoan, and viral antigens. Once formed, NO rapidly diffuses across both prokaryotic cell walls and eukaryotic cell membranes where it is then able to render cytocidal and cytostatic antimicrobial effects. There are several important mechanisms by which NO mediates antimicrobial activity involving the inhibition of cellular replication and energy production. NO interrupts DNA synthesis via inhibition of ribonucleotide reductase [131, 132]. Furthermore, NO avidly reacts with intracellular iron-containing molecules and via S-nitrosylation renders them fatally inactive [108, 133]. Also, NO in the presence of oxygen can form reactive oxygen intermediates, including peroxynitrite, which have been shown to be directly cytotoxic. To form peroxynitrite, NO reacts with superoxide anion generated by the respiratory burst of activated macrophages. Peroxynitrite then breaks down to produce the highly toxic products: hydroxyl radical and nitrogen dioxide [96]. In addition, NO causes autoribosylation of glyceraldehyde-3-phosphate dehydrogenase, which blocks glycolysis [63]. NO has also been shown to inhibit enzymes important in the Krebs cycle and of the electron transport chain [69, 134]. Thus, there are a number of NO-susceptible targets for antimicrobial activity. This is well characterized in the study of NO and malarial infestation.
The antimicrobial role of NO in the liver is best exemplified in malaria, an intracellular pathogen which accounts for over two million deaths per year world-wide. After being transmitted by the anopheles mosquito, malaria sporozoites penetrate hepatocytes and complete their life cycle by maturing into liver-staged schizonts. There they reproduce, many thousand times over, before bursting from the liver and initiating the symptomatic blood stage of the infection.
Green et al. demonstrated that P. berghei-infected hepatocytes in culture treated with IFN-gamma resulted in a significant decrease in the number of intracellular schizonts [135]. However, the protective effects of IFN-gamma are inhibited by L-NMMA [112]. In addition, Mellouk and coworkersreported the potential role of NO in anti-malarial immunity. Here, the typically successful immunization of mice against hepatic stages of P. berghei and P. yeolli with irradiated sporozoites was blocked by the simultaneous injection of either L-NMMA and anti-IFN-gamma antibodies [136]. Thus, NO appears to be an important recognition molecule in the development of anti-malarial immunity. Another mechanism for the anti-malarial effect of NO occurs during the blood-borne stage of the disease by NO diffusing into Plasmodium infected red-blood cells and complexing with cysteine or glutathione to form nitrosothiol groups which are highly toxic to the merozoites [137].
The anti-malarial effect of NO is also evident in human hepatocytes. First, human hepatocytes have preserved the capacity to produce high, sustained iNOS expression, whereas human monocytes and macrophages are unable to produce comparable levels of NO. This suggests that iNOS expression is well tolerated by human hepatocytes, and since many major infections have a liver component in which NO exerts an important antimicrobial effect, the capacity for iNOS expression has been conserved in the human liver. Whereas hepatocytes my be protected from injury, the same is not true for intracellular pathogens. Mellouk et al. demonstrated that IFN-gamma leads to elimination of the parasite Plasmodium falciparum in primary human hepatocyte cultures and that the anti-malarial activity is dependent on the cytotoxic molecule NO [136]. Interestingly, the parasite itself stimulates iNOS expression in human hepatocytes independently of added cytokines [138]. In addition, they were able to demonstrate that both spontaneous and IFN-gamma-induced inhibition of the malaria parasite could be increased with the addition of the NO synthase cofactors BH4 and sepiaterin [136]. These results indicate that under in vitro conditions, the parasite itself provides a signal that triggers induction of the NO pathway in hepatocytes, and NO formation in infected hepatocytes may be limited by cofactor availability. This may imply that NO formation may be increased in vivo and malaria could be more effectively treated by providing the cofactor for BH4 to travelers and people living in malaria-endemic countries. Since hepatocytes exhibit potent anti-malarial activity mediated via NO, these results suggest that cells may directly respond to parasite infection by iNOS expression to limit microbial growth.
CONCLUSIONS
Research to date has demonstrated that the regulation of iNOS is complex and is governed by transcriptional, post-transcriptional, translational, and post-translational mechanisms. These multiple levels of regulation lead to one to speculate that iNOS expression is tightly controlled. Since NO production has both beneficial and detrimental effects, understanding the molecular mechanisms that govern iNOS expression is critical to developing strategies to manipulate NO release in pathophysiologic conditions.
The important factors in determining the beneficial versus harmful effects of NO in the liver are the amount and duration of NO production. First, iNOS expression during acute inflammation and early sepsis is beneficial secondary to its protective effects in the liver whereby NO serves to maximize perfusion. In addition, NO prevents platelet aggregation, thrombosis, and neutralizes toxic oxygen radicals. Also, NO demonstrates antimicrobial properties and prevents neutrophil activation. iNOS expression in hepatocytes is also hepatoprotective during more severe insults. iNOS-derived NO induces the expression of Hsp70 and heme oxygenase, both categories of protective proteins. This level of NO production also mediates anti-apoptotic effects in the liver cells. Furthermore, while NO may play a role in the development of liver cancer in the setting of chronic liver inflammation, it also mediates anti-tumor activity when produced by inflammatory cells during the immune response against tumor cells.
The clinical application of modulation of NO function in a number of liver disease processes is promising. However, NO is a ubiquitous mediator, and as such, nonspecific inhibition or augmentation of its activity will lead to a number of derangements of homeostasis. The emergence of iNOS-selective inhibitors as well as NO-donors with cell-specific action is on the horizon. Further research of the role of NO in liver pathophysiology will provide powerful tools to manipulate a number of hepatic and gastrointestinal disorders.
REFERENCES
1.Moncada, S., and Higgs, E. A. (1991) Eur. J.
Clin. Invest., 21, 361-374.
2.Hibbs, J. B., Vavrin, Z., and Taintor, R. R.
(1987) J. Immun., 138, 550-565.
3.Palmer, R. M., Ferrige, A. G., and Moncada, S.
(1987) Nature, 327, 524-526.
4.Ignarro, L. J., Buga, G. M., Wood, K. S., Byrns, R.
E., and Chaudhuri, G. (1987) Proc. Natl. Acad. Sci. USA,
84, 9265-9269.
5.Billiar, T. R., and Curran, R. D. (1990) J.
Parenter. Enteral. Nutr., 14, S175-S180.
6.Stuehr, D. J., and Marletta, M. A. (1985) Proc.
Natl. Acad. Sci. USA, 82, 7738-7742.
7.Billiar, T. R., Curran, R. D., Steuhr, D. J., West,
M. A., Bentz, B. A., and Simmons, R. L. (1989) J. Exp. Med.,
169, 1467-1472.
8.Curran, R. D., Billiar, T. R., Steuhr, D. J.,
Hoffmann, K., and Simmons, R. L. (1989) J. Exp. Med.,
170, 1769-1774.
9.Curran, R. D., Billiar, T. R., Steuhr, D. J.,
Ochoa, J. B., Harbrecht, B. G., Flint, S. G., and Simmons, R. L. (1990)
Ann. Surg., 212, 462-471.
10.Geller, D. A., de Vera, M. E., Russell, D. A.,
Shapiro, R. A., Nussler, A. K., Simmons, R. L., and Billiar, T. R.
(1995) J. Immunol., 155, 4890-4898.
11.Geller, D. A., Nussler, A. K., Di Silvio, M.,
Lowenstein, C. L., Shapiro, R. A., Wang, S. C., Simmons, R. L., and
Billiar, T. R. (1993) Proc. Natl. Acad. Sci. USA, 90,
522-526.
12.Pittner, R. A., and Spitzer, J. A. (1992)
Biochem. Biophys. Res. Commun., 185, 430-435.
13.Freeswick, P. D., Wan, Y., Geller, D. A.,
Nussler, A. K., and Billiar, T. R. (1994) J. Surg. Res.,
57, 205-209.
14.Geller, D. A., Freeswick, P. D., Nguyen, D.,
Nussler, A. K., Di Silvio, M., Shapiro, R. A., Wang, S. C., Simmons, R.
L., and Billiar, T. R. (1994) Arch. Surg., 129,
165-171.
15.Gaillard, T., Mulsch, A., Klein, H., and Decker,
K. (1992) Biol. Chem. Hoppe. Seyler, 373, 897-902.
16.Knowles, R. G., Salter, M., Brooks, S. L., and
Moncada, S. (1990) Biochem. Biophys. Res. Commun., 172,
1042-1048.
17.Taylor, B., Kim, Y. M., Wang, Q., Shapiro, R. A.,
Billiar, T. R., and Geller, D. A. (1997) Arch. Surg., in
press.
18.Liu, Z. Z., Cui, S., Billiar, T. R., Dorko, K.,
Halfter, W., Geller, D. A., Michalopoulos, G., Beger, H. G., Albina,
J., and Nussler, A. K. (1996) J. Leuk. Biol., 60,
382-388.
19.Schmidt, H. H., Polack, J. S., Nakane, M.,
Gorsky, L. D., Forstermann, U., and Murad, F. (1991) Proc. Natl.
Acad. Sci. USA, 88, 365-369.
20.Spitzer, J. A. (1994) Hepatology,
19, 217-228.
21.Nussler, A., Di Silvio, M., Billiar, T. R.,
Hoffman, R. A., Geller, D. A., Selby, R., Madriaga, J., and Simmons, R.
L. (1992) J. Exp. Med., 176, 261-264.
22.Geller, D. A., Lowenstein, C. J., Shapiro, R. A.,
Nussler, A. K., Di Silvo, M., Wang, S. C., Nakayama, D. K., Simmons, R.
L., Snyder, S. H., and Billiar, T. R. (1993) Proc. Natl. Acad. Sci.
USA, 90, 3491-3495.
23.Wettstein, M., Gerok, W., and Haussinger, D.
(1994) Hepatology, 19, 641-647.
24.Pastor, C. M., Morris, S. M., and Billiar, T. R.
(1995) Am. J. Physiol., 269, G861-G866.
25.Lyons, C. R., Orloff, G. J., and Cunningham, J.
M. (1992) J. Biol. Chem., 267, 6370-6374.
26.Xie, Q., Cho, H. J., Calaycay, J., Mumford, R.
A., Swiderek, K. M., Lee, T. D., Ding, A., Troso, T., and Nathan, C.
(1992) Science, 256, 225-228.
27.Lowenstein, C. J., Glatt, C. S., Bredt, D. S.,
and Snyder, S. H. (1992) Proc. Natl. Acad. Sci. USA, 89,
6711-6715.
28.Wood, E. R., Berger, H., Sherman, P. A., and
Lapetina, E. G. (1993) Biochem. Biophys. Res.
Commun.,191, 767-774.
29.Nunokawa, Y., Ishida, N., and Tanaka, S. (1993)
Biochem. Biophys. Res. Commun., 191, 89-94.
30.Charles, I. G., Palmer, R. M. J., Hickery, M. S.,
Bayliss, M. T., Chubb, A. P., Hall, V. S., Moss, D. W., and Moncada, S.
(1993) Proc. Natl. Acad. Sci. USA, 90, 11419-11423.
31.Sherman, P. A., Laubach, V. E., Reep, B. R., and
Wood, E. R. (1993) Biochemistry, 32, 11600-11605.
32.Hokari, A., Zeniya, M., and Esumi, H. (1994)
J. Biochem., 116, 575-581.
33.Kozak, M. (1991) J. Cell Biol.,
115, 887-903.
34.Marsden, P. A., Schappert, K. T., Chen, H. S.,
Flowers, M., Sundell, C. L., Wilcox, J. N., Lamas, S., and Michel, T.
(1992) FEBS Lett., 307, 287-293.
35.Nakane, M., Schmidt, H. H. H. W., Pollock, J. S.,
Forstermann, U., and Murad, F. (1993) FEBS Lett., 316,
175-180.
36.Cho, H. J., Xie, Q.-W., Calaycay, J., Mumford, R.
A., Swiderek, K. M., Lee, T. D., and Nathan, C. (1992) J. Exp.
Med., 176, 599-604.
37.Chartrain, N., Geller, D. A., Koty, P. P.,
Sitrin, N. F., Nussler, A. K., Hoffman, E. P., Billiar, T. R.,
Hutchinson, N. I., and Mudgett, J. S. (1994) J. Biol. Chem.,
269, 6765-6772.
38.De Vera, M. E., Shapiro, R. A., Nussler, A. K.,
Mudgett, J. S., Simmons, R. L., Morris, S. M., Billiar, T. R., and
Geller, D. A. (1996) Proc. Natl. Acad. Sci. USA, 93,
1054-1059.
39.Xie, Q. W., Whisnan, R., and Nathan, C. (1993)
J. Exp. Med., 177, 1779-1784.
40.Lowenstein, C. J., Alley, E. W., Raval, P.,
Snowman, A. M., Snyder, S. H., Russell, S. W., and Murphy, W. J. (1993)
Proc. Natl. Acad. Sci. USA, 90, 9730-9734.
41.Geller, D. A., Nussler, A. K., Di Silvio, M.,
Lowenstein, C. J., Shapiro, R. A., Wang, S. C., Simmons, R. L., and
Billiar, T. R. (1993) Proc. Natl. Acad. Sci. USA, 90,
522-526.
42.De Vera, M. E., Taylor, B. S., Wang, Q., Shapiro,
R. A., Billiar, T. R., and Geller, D. A. (1997) Am. J. Phys., in
press.
43.Kleinert, H., Euchenhofer, C., Ihrig-Biedert, I.,
and Forstermann, U. (1996) Mol. Pharmacol., 49,
15-21.
44.De Vera, M. E., Wong, J., Zhou, J. Y., Tzeng, E.,
Wong, H., Billiar, T. R., and Geller, D. A. (1996) Surgery,
120, 144-149.
45.De Vera, M. E., Kim, Y. M., Wong, H. R., Wang,
Q., Billiar, T. R., and Geller, D. A. (1996) Hepatology,
24, 1238-1245.
46.Feinstein, D. L., Galea, E., Aquino, D. A., Li,
G. C., Xu, H., and Reis, D. J. (1996) J. Biol. Chem.,
271, 17724-17732.
47.Kim, Y. M., de Vera, M. E., Watkins, S. C., and
Billiar, T. R. (1997) J. Biol. Chem., 272, 1402-1411.
48.Liu, Z. Z., Cui, S., Billiar, T. R., Dorko, K.,
Halfter, W., Geller, D. A., Michalopoulos, G., Beger, H. G., Albina,
J., and Nussler, A. K. (1996) J. Leuk. Biol., 60,
382-388.
49.Forrester, K., Ambs, S., Lupold, S. E., Kapust,
R. B., Spillare, E. A., Weinberg, W. C., Felley-Bosco, E., Wang, X. W.,
Geller, D. A., Tzeng, E., Billiar, T. R., and Harris, C. C. (1996)
Proc. Natl. Acad. Sci. USA, 93, 2442-2447.
50.Shaw, G., and Kamen, R. (1986) Cell,
46, 659-667.
51.Caput, D., Beutler, B., Hartog, K., Thayer, R.,
Brown-Shimer, S., and Derami, A. (1986) Proc. Natl. Acad. Sci.
USA, 83, 1670-1674.
52.Han, J., Brown, T., and Beutler, B. (1990) J.
Exp. Med., 171, 465-475.
53.Pastor, C. M., Williams, D., Yoneyama, T.,
Hatakeyama, K., Naylor, E., and Billiar, T. R. (1996) J. Biol.
Chem.,271, 24534-24538.
54.Harada, T., Kagamiyama, H., and Hatakeyama, K.
(1993) Science, 260, 1507-1510.
55.Heinrich, P. C., Castell, J. V., and Andus, T.
(1990) Biochem. J., 265, 621-636.
56.West, M. A., Billiar, T. R., Mazuski, J. E.,
Curran, R. D., and Simmons, R. L. (1988) Arch. Surg.,
123, 1400-1405.
57.Billiar, T. R., Curran, R. D., West, M. A.,
Hofmann, K., and Simmons, R. L. (1989) Arch. Surg., 124,
1416-1420.
58.Curran, R. D., Ferrari, F. K., Kispert, P. H.,
Stadler, J., Stuehr, D. J., Simmons, R. L., and Billiar, T. R. (1991)
FASEB J., 5, 2085-2092.
59.Billiar, T. R., Curran, R. D., Ferrari, F. K.,
Williams, D. L., and Simmons, R. L. (1990) J. Surg. Res.,
48, 349-353.
60.Sax, H. C., Talamini, M. A., Hasselgren, P. O.,
Rosenblum, L., Ogle, C. K., and Fischer, J. E. (1988) J. Surg.
Res., 44, 109-116.
61.Frederick, J. A., Hasselgren, P. O., Davis, S.,
Higashiguchi, T., Jacob, T. D., and Fischer, J. E. (1993) Arch.
Surg., 128, 152-157.
62.Brass, E. P., and Vetter, W. H. (1993)
Pharmacol. Toxicol., 72, 369-372.
63.Horton, R. A., Leppi, E. D., Knowles, R. G., and
Titheradge, M. A. (1994) Biochem. J., 299, 735-739.
64.Molina y Vedia, L., McDonald, B., Reep, B.,
Brune, B., Di Silvio, M., Billiar, T. R., and Lapetina, E. G. (1992)
J. Biol. Chem., 267, 24929-24932.
65.Ou, J., Molina, L., Kim, Y. M., and Billiar, T.
R. (1996) Am. J. Physiol., 271, G621-G628.
66.Nathan, C. (1992) FASEB J., 6,
3051-3064.
67.Wood, K. S., and Ignarro, L. J. (1987) J.
Biol. Chem., 262, 5020-5027.
68.Billiar, T. R., Curran, R. D., Harbrecht, B. G.,
Stadler, J., Williams, D. L., Ochoa, J. B., Di Silvio, M., Simmons, R.
L., and Murray, S. A. (1992) Am. J. Physiol., 262,
C1077-C1082.
69.Saavedra, J. E., Billiar, T. R., Williams, D. L.,
Kim, Y. M., Watkins, S. C., and Keefer, L. K. (1997) J. Med.
Chem., in press.
70.Stadler, J., Billiar, T. R., Curran, R. D.,
Steuhr, D. J., Ochoa, J. B., and Simmons, R. L. (1991) Am. J.
Physiol., 260, C910-C916.
71.Kurose, I., Kato, S., Ishii, H., Fukumura, D.,
Miura, S., Suematsu, M., and Tsuchiya, M. (1993) Hepatology,
18, 380-388.
72.Ghezzi, P., Saccardo, B., Villa, P., Rossi, V.,
Bianchi, M., and Dinarello, C. S. (1986) Infect. Immun.,
54, 837-840.
73.Peterson, T. C., and Renton, K. W. (1986)
Immunopharmacology, 11, 21-28.
74.Khatsenko, O. G., Gross, S. S., Rifkind, A. B.,
and Vane, J. R. (1993) Proc. Natl. Acad. Sci. USA, 90,
11147-11151.
75.Stadler, J., Trockfeld, J., Schmalix, W. A.,
Brill, T., Siewert, J. R., Greim, H., and Doehmer, J. (1994) Proc.
Natl. Acad. Sci. USA, 91, 3559-3563.
76.Ochoa, J. B., Udekwu, A. O., Billiar, T. R.,
Curran, R. D., Cerra, F. B., Simmons, R. L., and Peitzman, A. B. (1991)
Ann. Surg., 214, 621-626.
77.Billiar, T. R., Curran, R. D., Harbrecht, B. G.,
Stuehr, D. J., Demetris, A. J., and Simmons, R. L. (1990) J. Leuk.
Biol., 48, 565-569.
78.Harbrecht, B. G., Billiar, T. R., Stadler, J.,
Demetris, A. J., Ochoa, J., Curran, R. D., and Simmons, R. L. (1992)
J. Leuk. Biol., 52, 390-394.
79.Harbrecht, B. G., Stadler, J., Demetris, A. J.,
Simmons, R. L., and Billiar, T. R. (1994) Am. J. Physiol.,
266, G1004-G1010.
80.Harbrecht, B. G., Billiar, T. R., Stadler, J.,
Demetris, A. J., Ochoa, J. B., Curran, R. D., and Simmons, R. L. (1992)
Crit. Care Med., 20, 1568-1574.
81.Szabo, C., Southan, G. J., and Thiemermann, C.
(1994) Proc. Natl. Acad. Sci. USA, 91, 12472-12476.
82.Ou, J., Carlos, T. M., Saavedra, J. E., Keefer,
L. K., Watkins, S. C., Harbrecht, B. G., and Billiar, T. R. (1997), in
press.
83.Bursch, W., Oberhammer, F., Jirtle, R. L.,
Askari, M., Sedivy, R., Grasl-Kraupp, B., Purchio, A. F., and
Schulte-Hermann, R. (1993) Br. J. Cancer, 67,
531-536.
84.Watanabe-Fukunaga, R., Brannan, C. I., Copeland,
N. G., Jenkins, N. A., and Nagata, S. (1992) Nature, 56,
314-317.
85.Leist, M., Gantner F., Jilg, S., and Wendel, A.
(1995) J. Immunol., 154, 1307-1316.
86.Albina, J. E., Cui, S., Mateo, R. B., and
Reichner, S. J. (1993) J. Immunol., 150, 5080-5085.
87.Lipton, S. A., Choi, Y. B., Pan, Z. H., Lei, S.
Z., Chen, H. S., Sucher, N. J., Loscalzo, J., Singel, D. J., and
Stamler, J. S. (1993) Nature, 364, 626-632.
88.Martin-Sanz, P., Diaz-Guerra, M. J. M., Casado,
M., and Bosca, L. (1996) Hepatology, 23, 1200-1207.
89.Bohlinger, I., Leist, M., Barsig, J., Uhlig, S.,
Tiegs, G., and Wendel, A. (1995) Hepatology, 22,
1829-1837.
90.Kim, Y. M., Talanian, R. V., and Billiar, T. R.
(1997) J. Biol. Chem., in press.
91.Kuo, P. C., and Slivka, A. J. (1994) Surg.
Res., 56, 594-600.
92.Arthur, M. J. P. (1988) J. Hepatol.,
6, 125-131.
93.Kanner, J., Harel, S., and Granit, R. (1991)
Arch. Biochem. Biophys., 289, 130-136.
94.Arthur, M. J. P., Bentley, I. S., Tanner, A. R.,
Kowalski-Saunders, P., Millward-Sadler, G. H., and Wright, R. (1985)
Gastroenterology, 89, 1114-1122.
95.Kim, Y. M., Bergonia, H., and Lancaster, J. R.
(1995) FEBS Lett., 374, 228-232.
96.Beckman, J. S., Beckman, T. W., Chen, J.,
Marshall, P. A., and Freeman, B. A. (1990) Proc. Natl. Acad. Sci.
USA, 87, 1620-1624.
97.Bautista, A. P., and Spitzer, J. J. (1994) Am.
J. Physiol., 266, G783-G788.
98.Ma, T. T., Ischiropoulos, H., and Brass, C. A.
(1995) Gastroenterology, 108, 463-469.
99.Christopher, T. A., Ma, X., and Lefer, A. M.
(1994) Shock, 1, 19-24.
100.Jaeschke, H., Schini, V. B., and Farhood, A.
(1992) Life Sci., 50, 1797-1804.
101.Liu, P., Yin, K., Yue, G., and Wong, P. Y. K.
(1996) J. Inflamm., 46, 144-154.
102.Harbrecht, B. G., Wu, B., Watkins, S. C.,
Marshall, H. P., Jr., Peitzman, A. B., and Billiar, T. R. (1995)
Shock, 4, 332-337.
103.Kobayashi, H., Nonami, T., Kurokawa, T.,
Takeuchi, Y., Harada, A., Nakao, A., and Takagi, H. (1995) J. Surg.
Res., 59, 772-779.
104.Wang, Y., Mathews, W. R., Guido, D. M.,
Farhood, A., and Jaeschke, H. (1995) Shock, 4,
282-288.
105.Ohshima, H., Brouet, I. M., Bandaletova, H.,
Adachi, S., Oguchi, S., Iida, S., Kurashima, Y., Morishita, Y.,
Sugimura, T., and Esumi, H. (1992) Biochem. Biophys. Res.
Commun., 187, 1291-1297.
106.Liu, R. H., Jacob, J. R., Hotchkiss, J. H.,
Cote, P. J., Gerin, J. L., and Tennant, B. C. (1994)
Carcinogenesis, 15, 2875-2877.
107.Ohshima, H., and Bartsch, H. (1994) Mutat.
Res., 305, 253-264.
108.Keefer, L. K., and Wink, D. A. (1996) Adv.
Exp. Med. Biol., 387, 177-185.
109.Stuehr, D. J., and Nathan, C. T. (1989) J.
Exp. Med., 169, 1543-1555.
110.Fukumura, D., Yonei, Y., Kurose, I., Saito, H.,
Ohishi, T., Higuchi, H., Miura, S., Kato, S., Kimura, H., Ebinuma, H.,
and Ishi, H. (1996) Hepatology, 24, 141-149.
111.Epstein, M. (1990) in Hepatology (Zakim,
D., and Boyer, T. D., eds.) Saunders, Philadelphia, pp. 493-512.
112.Korthuis, R. J., Benoit, J. N., Kvietys, P. R.,
Townsley, M. I., Taylor, A. E., and Granger, D. N. (1985) Am. J.
Physiol., 249, H827-H833.
113.Lumsden, A. B., Henderson, J. M., and Kutner,
M. H. (1988) Hepatology, 8, 232-236.
114.Triger, D. R., Boyer, T. D., and Levin, J.
(1978) Gut, 19, 935-939.
115.Groszmann, R., Kotelanski, B., Cohn, J. N., and
Khatri, I. M. (1972) Am. J. Med., 53, 715-722.
116.Hori, N., Okanoue, T., Mori, T., Kashima, K.,
Nishimura, M., Nanbu, A., Yoshimura, M., and Takahashi, H. (1996)
Clin. Exp. Pharmacol. Physiol., 23, 30-35.
117.Miyase, S., Fujiyama, S., Chikazawa, H., and
Sato, T. (1990) Gastroenterol. Jpn., 25, 356-362.
118.Niederberger, M., Gines, P., Tsai, P., Martin,
P. Y., Morris, K., Weigert, A., McMurtry, I., and Schrier, R. W. (1995)
Hepatology, 21, 1625-1631.
119.Radomski, M. W., Palmer, R. M. J., and Moncada,
S. (1990) Proc. Natl. Acad. Sci. USA, 87,
10043-10047.
120.Pizcueta, P., Pique, J. M., Bosch, J., Whittle,
B. J. R., and Moncada, S. (1991) Gastroenterology, 100,
A785.
121.Niederberger, M., Martin, P. Y., Gines, P.,
Morris, K., Tsai, P., Xu, D. L., McMurtry, I., and Schrier, R. W.
(1995) Gastroenterology, 109, 1624-1630.
122.Oshita, M., Takei, Y., Kawano, S., Hijioka, T.,
Masuda, E., Goto, M., Nishimura, Y., Nagai, H., Iio, S., Tsuji, S.,
Fusamoto, H., and Kamada, T. (1994) Hepatology, 20,
961-965.
123.Vouldoukis, I., Riveros-Moreno, V., Dugas, B.,
Ouaaz, F., Becherel, P., Debre, P., Moncada, S., and Mossalayai, M. D.
(1995) Proc. Natl. Acad. Sci. USA, 92, 7804-7808.
124.Woods, L. F., Wood, J. M., and Gibbs, P. A.
(1981) J. Gen. Microbiol., 125, 399-406.
125.Granger, D. L., Hibbs, J. B., Jr., Perfect, J.
R., and Durack, D. T. (1988) J. Clin. Invest., 81,
1129-1136.
126.James, S. L., and Glaven, J. (1989) J.
Immunol., 143, 4208-4212.
127.Vincendeau, P., Daulouede, S., Veyret, B.,
Darde, M. L., Bouteille, B., and Lemesre, J. L. (1992) Exp.
Parasitol., 75, 353-360.
128.Munoz-Fernadez, M. A., Fernandz, M., and
Fresno, M. (1992) Immunol. Lett., 33, 35-40.
129.Flesch, I. E., Hess, J. H., and Kaufmann, S. H.
(1994) Int. Immunol., 6, 1751-1757.
130.Vouldoukis, I., Mazier, D., Debre, P., and
Mossialayi, M. D. (1995) Res. Immunol., 146, 689-692.
131.Lepoivre, M., Chenais, B., Yapo, A., Lemaire,
G., Thelander, L., and Tenu, J. P. (1990) J. Biol. Chem.,
265, 14143-14149.
132.Kwon, N. S., Stuehr, D. J., and Nathan, C. F.
(1991) J. Exp. Med., 174, 761-767.
133.Hibbs, J. B., Jr., Taintor, R. R., Vavrin, Z.,
and Rachlin, E. M. (1988) Biochem. Biophys. Res. Commun.,
157, 87-94.
134.Spence, J. T., Merrill, M. J., and Pitot, H. C.
(1981) J. Biol. Chem., 256, 1598-1603.
135.Green, S. J., Mellouk, S., Hoffman, S. L.,
Meltzer, M. S., and Nacy, C. A. (1990) Immunnol. Lett.,
25, 15-19.
136.Mellouk, S., Hoffman, S. L., Liu, Z. Z., de la
Vega, P., Billiar, T. R., and Nussler, A. K. (1994) Infect.
Immun., 62, 4043-4047.
137.Rockett, K. A., Awburn, M. M., Aggarwal, B. B.,
Cowden, W. B., and Clark, I. A. (1992) Infect. Immun.,
60, 3725-3730.
138.Nussler, A., Renia, L., Pasquetto, V., Miltgen,
F., Matile, H., and Mazier, D. (1993) Eur. J. Immunol.,
23, 882-887.