REVIEW: Dinitrosyl Iron Complexes and S-Nitrosothiols Are Two Possible Forms for Stabilization and Transport of Nitric Oxide in Biological Systems
A. F. Vanin
Institute of Chemical Physics, Russian Academy of Sciences, ul. Kosygina 4, Moscow, 117977 Russia; fax: (095) 938-2156; E-mail: mikoyan@center.chph.ras.ru
Received February 18, 1998
The physicochemical properties, mechanisms of synthesis and decomposition of dinitrosyl iron complexes (DNICs) with thiol-containing ligands and of S-nitrosothiols (RS-NO), and the potential role of these compounds in storage and transport of NO in biological systems are reviewed. Special attention is given to the phenomenon of mutual transformation of DNIC and RS-NO catalyzed by Fe2+. Each Fe2+ binds two neutral NO molecules in the DNICs, catalyzes their mutual oxidation--reduction with formation of nitrous oxide and nitrosonium ions appearing in the DNICs. These ions S-nitrosate thiol-compounds with RS-NO formation. Fe2+ binds two RS-NO molecules and catalyzes their mutual oxidation--reduction followed by decomposition of the resulting molecules. Mutual conversion of DNICs and RS-NO regulated by iron, thiol, and NO levels is suggested to provide NO transport in cells and tissues.
KEY WORDS: nitric oxide, dinitrosyl iron complexes, S-nitrosothiols, cells and tissues
Abbreviations: cys-NO) S-nitrosocysteine; DNIC) dinitrosyl iron complexes; DETC) diethyldithiocarbamate anion; EDRF) endothelium-derived relaxing factor; EPR) electron paramagnetic resonance; GSH) reduced glutathione; NAC) N-acetyl-L-cysteine; NAP) N-acetyl-DL-penicillamine; NO) nitric oxide; NOS) nitric oxide synthase; RS-NO) S-nitrosothiols; SNAC) S-nitroso-N-acetyl-L-cysteine; SNAP) S-nitroso-N-acetyl-DL-penicillamine; HFS) hyperfine structure.
Endogenous formation of one of the simplest chemical compounds, nitric
oxide (NO), in animals and humans is now well recognized. NO is
enzymatically synthesized from L-arginine; it plays an important role
as a regulator of metabolism in cells and tissues [1-5]. NO functions as the main
cytostatic/cytotoxic effector of the cellular immunity system [6-8]. NO exerts both autocrine and
paracrine actions, i.e., being synthesized in certain cells it can
influence metabolic processes in these and adjacent cells. The latter
means that in spite of high chemical reactivity, NO molecules can be
transported to a distance several-fold exceeding cell sizes [9]. During this transportation free NO molecules can
be caught by various endogenous scavengers, for example, by hemoglobin;
hemoglobin catalyzes NO oxidation by oxygen. Superoxide ions can also
effectively oxidize NO. These factors can significantly attenuate or
even abolish the paracrine effect of free NO molecules. So, only
reversible incorporation of NO into compounds that can transport it
from the donor cells to target cells can prevent or diminish these
processes. Experiments confirmed that nature has chosen this mechanism.
This may be illustrated by so-called "endothelium-derived relaxing
factor” (EDRF) of blood vessels [10]. In 1987
Moncada et al. showed that EDRF contains NO and it is this particular
component which is responsible for the vasodilator activity of EDRF.
Moreover, it was demonstrated that the efficacy of free NO added to
isolated vessels in amounts equal to that found in EDRF was identical
to the efficacy of EDRF. It was thus concluded that EDRF and NO are
identical [11]. However, this result was not
confirmed in subsequent studies [12-14]. The vasodilator activity of EDRF (expressed per
amount of NO detected in EDRF) was one order of magnitude more
effective than that of free NO. This suggested that EDRF is a
nitroso-compound rather free NO. S-Nitroso-cysteine (cys-NO) which
protected its NO against oxidation and released it during contact with
smooth muscle cells was a good candidate for EDRF. Comparing EDRF and
NO capacities in formation of dinitrosyl iron complexes (DNICs), we
came to the same conclusion [15].
The choice of cys-NO as a candidate for EDRF was due to their almost identical efficacy of the vasodilator effect (with respect to the amount of NO in them) [12]. We proposed DNICs with thiol-containing ligands (cysteine or glutathione, GSH) as another candidate for EDRF [16]. It was shown that NO incorporation effectively protected them against the oxidizing effect of superoxide, and this resulted in a significant shift of the dose response curve of the dependence of isolated vessel relaxation on vasodilator dose to the left [17]. Vasodilator effects of DNIC with cysteine and cys-NO were equally effective [12, 17].
The involvement of NO produced from L-arginine in animal cells and tissues into the formation of both S-nitrosothiols (RS-NO) and DNICs with thiol-containing ligands has been demonstrated in numerous studies [18-26]. This fact together with data on the physicochemical and functional properties of RS-NO and DNIC suggest that these compounds can function in NO transportation not only in vessels, but also in other tissues, acting as forms for NO stabilization in animals and humans which provide NO transport over various cells and tissues. The present review is devoted to validation of this suggestion.
DNIC: PHYSICOCHEMICAL PROPERTIES, SYNTHESIS, AND
DECOMPOSITION
More than 30 years ago DNICs were found in animal tissues and microorganisms by their characteristic EPR signal, signal 2.03 according to its mean of the axially symmetrical tensor of the g-factor (see the figure) [27-29]. Using only this indicator it became possible to identify the nature of these paramagnetic centers and to study mechanisms of their formation in biological systems. Parameters of their EPR signal completely coincided with the EPR signal of frozen aqueous solutions of DNIC with cysteine or with GSH [30, 31] (the figure). This coincidence was not accidental, and it provided decisive information for identification of the paramagnetic centers. Treatment of these centers and low-molecular-weight DNICs with agents which bound iron ions and thiol groups caused loss of both types of paramagnetic centers. Their contacts with ethylxanthogenate resulted in formation of iron complexes containing one NO molecule, paramagnetic mononitrosyl iron complexes with ethylxanthogenate [29]. It was concluded that the paramagnetic centers responsible for signal 2.03 represent DNICs with thiol ligands [32-34].
This conclusion was confirmed by experiments where isolated animal tissues and microorganisms were treated with gaseous nitric oxide or sodium nitrite as a source of NO. This caused formation of paramagnetic centers giving signal 2.03 [33, 35]. Moreover, a diet with increased nitrite content also led to formation of these centers in animal organs and tissues in vivo [36-41]. Convincing evidence for the presence of iron in these centers was obtained by incorporation of 57Fe isotope, which was added into the drinking water together with sodium nitrite. The presence of this iron isotope caused some broadening of signal 2.03 in tissues of these animals which was indicated by the appearance of hyperfine structure (HFS) from the 57Fe nucleus characterized by spin I = 1/2 [38, 39]. Such transformation was also observed for the EPR signal of frozen DNICs with low-molecular-weight thiols during incorporation of 57Fe isotope instead of the natural abundance isotope (56Fe), the latter not possessing nuclear spin [41, 42] (the figure).
In contrast to low-molecular-weight analogs, the anisotropic shape of the EPR signal for DNIC in biological systems remained unchanged as the temperature is changed from 77 K to room temperature. At room temperature low-molecular-weight DNIC give a narrow symmetrical EPR signal at g = 2.03 with isotropic 13-component HFS. Incorporation of 57Fe into these complexes causes the appearance of an isotropic doublet shown by hyperfine interaction of an unpaired electron with the nucleus of this isotope [41, 42] (the figure). Narrowing of the EPR signal of low-molecular-weight DNIC as the temperature is increased is obviously due to the rapid mobility of these components at room temperature, which leads to averaging of the anisotropy of the g-factor and HFS. Lack of such averaging for signal 2.03 in biological systems suggests that the DNICs bind to proteins. This was confirmed by determining the EPR signal of DNICs bound to thiol-containing groups of proteins: actomyosin, aldolase, etc. At room temperature these complexes give the same shape of signal as that of frozen solutions [30, 41].a) Signal 2.03 in rabbit liver (1), yeast (2), and in frozen solution of DNIC with cysteine (3) (recorded at 77 K). b) Signal 2.03 in mouse liver obtained after one week treatment with nitrite and 57Fe-citrate (1) or nitrite and 56Fe-citrate (2) administered in drinking water; 3-6) the EPR signal of DNICs with 57Fe (3, 5) or 56Fe (4, 6). Recorded at 77 K (1-4) or at room temperature (5, 6).
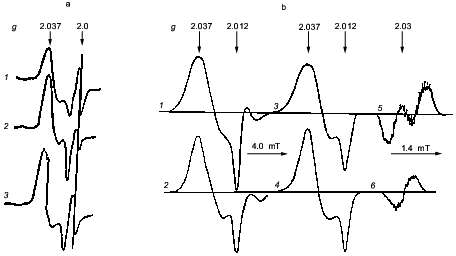
HFS analysis of the EPR signal of DNIC with cysteine done in 1965 [42] showed that these complexes contained molecules of cysteine and NO in pairs. The HFS appears as a results of unpaired electron interaction with the nitrogen nuclei of NO ligands present in the form of nitrosonium ions (NO+) and four protons of methylene groups of two cysteine ligands. Complete coincidence of DNIC with cysteine (at low temperature) and signal 2.03 in biological systems suggests that protein DNICs have the same composition. Paramagnetic DNICs can also appear in proteins containing only one thiol group, as for example in bovine or human serum albumin. In contrast to ordinary complexes they are characterized by lower (rhombic) symmetry: these complexes give an EPR signal with three different g-factor values [43]. This decrease in symmetry can be explained by incorporation into DNIC of a ligand rather than the thiol group of a protein, probably a histidine residue. Contact of this protein complex with low-molecular-weight thiols, cysteine or glutathione, causes reversible transformation of the DNIC: the histidine ligand is substituted by the low-molecular-weight thiol. The shape changes of the EPR signal indicate that DNIC, being protein bound, becomes axially symmetrical. Thus, it is possible that in cells and tissues giving signal 2.03 with axially symmetrical tensor of the g-factor of DNIC can contain two or one protein thiol residue. In the latter case the low-molecular-weight thiol is incorporated into the DNIC.
Formation of DNIC with low-molecular-weight thiols occurs at neutral and alkaline pH values when thiol solution and bivalent iron are mixed in an atmosphere of NO [30, 41, 42]. DNICs can exist in two forms, monomeric (paramagnetic) and dimeric (diamagnetic) [41, 44]. The first appears at concentration ratio thiol/Fe >10, whereas the second is formed when this ratio does not exceed 2. Depending on the thiol concentration these forms are interconvertible (Scheme 1):
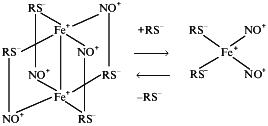
Scheme 1
It is suggested that the interaction of ionized thiol- and NO+-ligands and pairing of initially unpaired electrons mainly localized on iron atoms stabilize the dimer. The dimeric DNIC form is more stable than the monomeric form. DNICs bound to protein exist only as monomeric (paramagnetic) forms.
Formation of dimeric DNICs with cysteine or glutathione does not require pre-ionization of the thiol groups. At neutral pH, when only a small proportion of free cysteine or GSH is ionized [45], they are completely incorporated into the DNICs. Even at this pH the interaction of thiols with Fe(NO+)2 groups appears to be sufficient for their deprotonation and iron binding into the DNICs. However, conversion of dimeric DNICs into the monomeric forms requires increased concentration of pre-ionized free thiols (RS-). In the case of the DNIC with cysteine at neutral pH this requirement is achieved by 20-40-fold excess of free cysteine concentration over the iron concentration. In the case of the DNIC with GSH such excess of GSH is insufficient for the conversion of dimeric DNIC into the monomeric form. Conversion of the major part of dimeric DNIC with GSH into the paramagnetic monomers also requires pH increase up to 10-11.
Most researchers believe that DNICs with thiol-containing ligands represent low-spin complexes with effective electron spin S = 1/2. The unpaired electron is mainly localized on the dz2 iron orbital [46-49]. The electron configuration of the iron atom in DNICs is still is a matter of discussion. Some researches (including our group) suggest that the iron state can be described by d7 configuration [46, 47]. Others believe that the EPR parameters of DNICs can be best explained by electron configuration d9 [48, 49]. Consequently, taking into consideration the low-spin state of DNICs, the first group of scientists proposed a quadrate-planar DNIC spatial structure, whereas the second group of scientists believe that this represents a tetrahedral structure. Experimental data support both suggestions. X-Ray structural studies of crystals of DNICs with some low-molecular-weight thiols (other than cysteine or glutathione) revealed tetrahedral structure [50, 51], supporting the second suggestion. However, from iron electron configuration d9 typical for tetrahedral structure of DNICs it is hard to imagine that these complexes can accept two more electrons with their localization on the iron atom. Nevertheless this was experimentally observed in the reaction with a strong reductant, sodium dithionite [44, 52]. Such accepting behavior well corresponds to the iron electron configuration d7 and transition of iron into its d9 state.
DNICs formed in the reaction of neutral NO molecules with Fe2+ ions initially represent diamagnetic complexes with iron electron configuration d8 (six d-electrons from Fe2+ and two electrons from upper orbitals of two NO molecules). Transition of the complex into the paramagnetic state requires either one electron acceptance and transition into d9 or electron donation and transition into d7. The first process is less probable because it is hard to explain the origin of agents capable of one electron reduction of the DNIC in this system. One-electron oxidation of DNIC seems more probable. Formation of paramagnetic DNICs with various anionic ligands is accompanied by formation of equimolar quantities of nitrous oxide, N2O [53], i.e., by reduction of NO molecules into NO-. These ions (nitroxyl ions) bind protons, thus forming nitroxyl molecules which disproportionate with the formation of water and nitrous oxide molecules. In this connection the scheme of oxidation of the initial DNICs with electron configuration d8 to d7 was proposed [54]. A proton approaching one neutral NO molecule in the initial DNIC provokes electron transfer to it from the second NO molecule, the latter being converted into nitrosonium ion (NO+). In turn NO- binds a proton and leaves the complex as HNO and forms nitrous oxide. Its position in the complex is occupied by a neutral NO molecule which passes its electron to iron with formation of the paramagnetic DNIC with the structure (RS-)2Fe+(NO+)2:
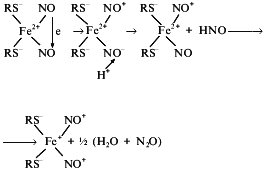
Scheme 2
Thus, it is suggested that mutual oxidation--reduction of NO occurs due to the approaching orientation to the iron atom and reaction of NO- with a proton. Actually, oxidation of some NO molecules to NO+ promotes reduction of other NO molecules to nitrous oxide (N2O).
Redox potentials of couples NO+/NO and NO/NO- (1.21 and 0.39 V, respectively [55]) do not seem to be able to provide mutual oxidation--reduction of NO molecules in DNIC. However, since NO reduction to NO- is accompanied by formation of nitroxyl (HNO) molecules dismutating with formation of a chemically inactive compound, nitrous oxide, there are some grounds for Scheme 2. The high redox potential of the couple NO/N2O (1.28 V) [55] also supports Scheme 2.
Some authors who detected N2O DNIC formation suggested that Fe2+ acts as NO reductant in these complexes [53]. However, gamma-resonance spectroscopy of iron incorporated into DNICs during their synthesis in the reaction of NO with Fe2+ revealed that all added iron could be incorporated into the DNIC [44]. A possible role of thiols as NO reductants suggested that their oxidation would diminish the synthesis of dimeric DNIC; however, this was not confirmed in experiments [44].
A proposed mechanism of paramagnetic DNIC formation can well explain recent data on the preferential formation of nitrous oxide, i.e., NO- anions and nitrite anions in solution of isolated NO-synthase (NOS) [56]. We interpret these results as the presence of iron contamination in this enzyme solution. Iron binds two neutral NO molecules which are converted into NO- and NO+ during electron transfer between them. Reactions of these ions with protons and hydroxyl ions result in formation of N2O and NO2-, respectively. This speculation can be easily checked by studying the influence of specific iron chelators in N2O formation in solution of NO-synthetase. Such experiments have already been carried out. It was shown [57] that on addition of the iron chelator N-methyl-D-glutamyldithiocarbamate to a solution of neuronal NOS, a significant proportion of the product was neutral NO molecules.
Spectroscopic studies of DNIC in infrared region revealed the existence of both NO-ligands in these complexes as nitrosonium ions [49]. During equilibrium decomposition of the complex one of these ions is converted into neutral NO molecule again in accordance with Scheme 3:
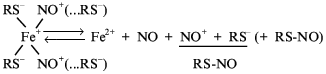
Scheme 3
This conversion was detected by the characteristic vasodilatory activity of DNIC diminished by hemoglobin molecules and also by DNIC-induced activation of guanylate cyclase [15, 44, 58]. The second nitrosonium ion released from DNIC causes S-nitrosylating activity of DNIC which is considered below.
Thus, consideration of the physicochemical properties of DNICs shows that they can play an important role in conversions and functioning of NO in living systems. First, they can act as storage and transport forms of NO. Second, at physiological pH they can provide conversion of neutral NO molecules into nitroso ions, stabilize these ions, and transport them in cells and tissues. This property of DNICs determines their interrelationship with the other possible form of stabilization of NO and its redox derivatives with RS-NO. According to Scheme 3, RS-NO can appear in the reaction of thiols with NO+ which can proceed either after release of these ions from DNIC or after equilibrium decomposition of DNIC accompanied by RS-NO release. Scheme 3 suggests involvement of RS-NO in the equilibrium with DNIC which apparently determines the stability of the latter. In fact, the high stability of the DNIC with GSH correlates with the high stability of S-nitrosoglutathione (GS-NO) and low stability of the DNIC with cysteine corresponds to low stability of cys-NO [44].
S-NITROSOTHIOLS: THEIR FORMATION AND DECOMPOSITION AT NEUTRAL
("PHYSIOLOGICAL") pH
In contrast to DNIC, RS-NO or thionitrites appeared as the result if proton substitution in SH-group for nitrosonium ion are diamagnetic. They are characterized by two absorbance bands at 338-340 and 540-543 nm with molar absorbance coefficients 930 and 22 M-1·cm-1, respectively. The physicochemical properties of these compounds are well studied (see for review [59, 60]). However, the problem of formation and decomposition of these compounds at neutral pH still requires solution.
Decomposition of S-nitrosothiols at neutral pH. The rate of RS-NO decomposition at neutral pH depends on the nature of its thiol. The half-time for cys-NO decomposition in aqueous solution at neutral pH does not exceed 1 min. It sharply increased (up to a few days) in the case of S-nitroso-derivative of the same amino acid during its incorporation into tripeptide (GSH) or proton substitution in the amino group for acetyl radical with formation of N-acetyl-L-cysteine (NAC). The half-time for decomposition of S-nitrosylated derivative of homocysteine, differing from cysteine by additional methylene group near the sulfur atom, is 1 h [61].
Redox agents destabilize RS-NO. One-electron reduction resulted in their decomposition with the release of thiol and neutral NO molecules. One-electron oxidation released thiyl radicals which rapidly form corresponding disulfides and nitrosonium ions, which are readily hydrolyzed with nitrite formation [14, 62]. The latter is also formed during the action of heavy metal ions on RS-NO, e.g., mercury and apparently copper ions displacing nitrosonium ions from these compounds due to higher affinity for thiols [63].
Another mechanism provides RS-NO decomposition by bivalent iron ions. The iron content in biological systems significantly exceeds the content of other metals [64]. In animal cells and tissues the iron content is two orders of magnitude more than that of copper, the other widely distributed metal. Other metals present in biological systems are in incomparably lower concentrations. The addition of selective iron chelators (desferal and o-phenanthroline) to solutions of RS-NO sharply increased the stability, for example, of the most unstable RS-NO, cys-NO [54, 65]. This suggests a destabilizing effect of iron ions on RS-NO. However, these experiments did not allow the conclusion that only binding of these chelators with iron ions presented as contaminants in chemicals used (thiols, buffer solution or accompanying salts) provided RS-NO stabilization. Selective (preferential) binding of iron ions by these chelators due to their higher affinity for iron ions does not mean that they are unable to bind other metals as well. For example, o-phenanthroline or 2,9-dimethyl-1,10-phenanthroline also bind copper ions [66]. So, it was possible such binding of contaminant copper ions could stabilize cys-NO. More convincing evidence for destabilization of RS-NO by iron ions was obtained in experiments where bivalent iron was added to cys-NO solution. Such experiments became possible after addition of millimolar (and higher) amounts of cysteine to the solution. This stabilized its S-nitroso-derivative and increased the half-time of decomposition from 30 sec (in the absence of cysteine) to 0.5 h (in the presence of cysteine) [65]. Under these conditions the addition of 10 and more micromoles of FeSO4 to millimolar solution of S-nitrosocysteine led to rapid decomposition of this RS-NO with formation of DNIC containing all of the added iron. If the cysteine concentration exceeded 10-fold the iron concentration, the paramagnetic (monomeric) form of DNIC predominated. If the ratio of cysteine and iron concentrations was lower, the diamagnetic (dimeric) form of these complexes predominated [54, 65].
The formation of a DNIC in cys-NO solution could be also due to a secondary reaction of Fe2+ binding with neutral NO molecules released from cys-NO. In this case NO scavengers (e.g., hemoglobin) would block or at least attenuate this process. However, addition of hemoglobin equimolar to the Fe2+ concentration did not influence the DNIC formation in a solution of cys-NO and cysteine within 1-2 min. It was thus concluded [54] that DNIC formation is due to the direct reaction between cys-NO with Fe2+ involving cysteine, possibly via the following mechanism (Scheme 4):
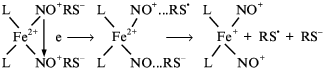
Scheme 4
Incorporation of two cys-NO molecules into the high-spin complex with iron is suggested to provide electron transfer between these molecules, i.e., their mutual oxidation--reduction destabilizing these compounds [14]. The oxidized cys-NO molecule is decomposed into nitrosonium ions and thiyl radical, whereas the reduced molecule produces neutral NO and the anionic form of the thiol. Thiyl radical can bind thiol with subsequent oxidation of the adduct to disulfide. The latter can also appear during recombination of thiyl radicals. Nitrosonium ion and NO molecule remain bound to iron. They form with it and thiol ligands the DNIC with iron electron configuration d7. According to chemical equilibrium between these complexes and their constituents (Scheme 3), the resultant DNIC provides release of neutral NO molecules and nitrosonium ions into solution.
Nitrosonium ions are obviously released into solution as the thiol-bound form, cys-NO. Interacting with iron ions these RS-NO are then decomposed with DNIC formation accompanied by release of NO and RS-NO (one molecule each from each complex), etc. According to Scheme 5, the decomposition of cys-NO and other RS-NO catalyzed by protein is terminated by release of neutral NO molecules into solution and accumulation of disulfides in solution:
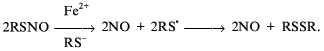
Scheme 5
Thus, according to this scheme Fe2+ catalyzes RS-NO decomposition via a homolytic mechanism. Numerous data suggest that this is the preferential mechanism for most RS-NO [60, 61, 67]. Decomposition of these compounds via heterolytic mechanism
Scheme 6
as a rule is realized in S-transnitrosylation reaction in which nitrosonium ion is transferred from one thiol to another:
Scheme 7
Copper ions can also catalyze RS-NO decomposition at neutral pH. This decomposition can be explained by copper iron binding (apparently as Cu+) with one RS-NO molecule with its subsequent decomposition via the homolytic mechanism [68, 69]. As catalysts of this process ions of contaminant copper (as contaminant iron) induce decomposition of such unstable RS-NO as cys-NO. Nevertheless, ions of contaminant iron plays the major role in this decomposition. At concentrations which do not exceed 50 µM the decomposing effects of iron and copper ions on cys-NO are equal [54]. If we take into consideration that in many commercial chemicals iron contamination significantly exceeds copper contamination, it is evident that ions of contaminant iron are mainly responsible for decomposition of cys-NO and, possibly, other RS-NO.
In accordance with Scheme 4, the RS-NO decomposition catalyzed by iron ions can proceed without free thiols. Other anionic compounds (L) such as buffer ions, halogens, phosphates, and finally, water molecules can be ligands of iron ions. The requirement for RS-NO decomposition is that these compounds can provide binding of two RS-NO molecules over a time interval sufficient for mutual redox conversion of these molecules. Such process leads to formation of relatively unstable DNIC with ligands of non-thiol nature. This instability is obviously due to rapid disappearance of nitrosonium ions released from DNIC in accordance with equilibrium process given in Scheme 3. Nitrosonium ions are hydrolyzed with formation of nitrite anions, and this leads to rapid DNIC decomposition and release of Fe2+ which binds RS-NO again with formation of rapidly decomposing DNIC. This finally leads to rapid disappearance of RS-NO.
Such a mechanism is probably responsible for cys-NO decomposition in the absence of cysteine. In this case cys-NO stabilization can be achieved by adding iron chelators. Similar results can also be obtained by adding cysteine. The resultant DNIC with cysteine (Scheme 4) is more stable than DNIC with non-thiol ligands (L). This fact is well documented in the literature [42, 46, 48]. Increase of DNIC stability is obviously due to NO+ stabilization via its binding to cysteine. The resultant cys-NO molecules maintain equilibrium of DNIC with its components (Scheme 3) providing DNIC stability. At micromolar (contaminant) concentration of iron release of Fe2+ in solution and their destructive effect on cys-NO are unlikely. Marked decomposition of cys-NO in the solution begins when the concentration of Fe2+ exceeds 10 µM.
In contrast to cys-NO, S-nitrosoglutathione (GS-NO) is destroyed by Fe2+ in the absence of GSH. Apparently, due to steric difficulties, two molecules of this tripeptide cannot form stable complexes with Fe2+ and non-thiol ligands which might provide mutual oxidation--reduction of GS-NO molecules. This process is initiated by GSH addition into solution, and addition of not less than 50 µM Fe2+ significantly accelerates it. This can illustrated by the following data [70] when 0.1 mM Fe2+ was added to 1 mM GS-NO containing 5 mM GSH, pH 7.2. After 10 min the absorbance spectrum besides GS-NO absorbance at 340 nm was characterized by appearance of bands at 310 and 360 nm typical for dimeric form of DNIC with GSH [71] which included all added iron ions. Due to time-dependent attenuation of GS-NO absorbance the resolution of these bands improved because of GS-NO decomposition with constant formation of DNIC. This process terminated within 1 h and during next 20-30 min the DNIC was completely destroyed.
In similar experiments with S-nitroso-N-acetyl-DL-penicillamine (SNAP) or S-nitroso-N-acetyl-L-cysteine (SNAC) [70] iron did not influence decomposition of these S-nitrosothiols in the presence of N-acetyl-DL-penicillamine or NAC, respectively. Apparently, iron ion chelation by acetyl-acetonate group of these thiols hampered the proceeding of this process via Scheme 4. Copper ions in the presence of corresponding thiols destroyed GS-NO, SNAP, or SNAC with equal efficacy. The latter suggests differences in mechanisms of RS-NO decomposition induced by copper and iron ions. In the experiments with GS-NO this difference consists in the route of nitric oxide. During iron-dependent GS-NO decomposition NO is not immediately released into solution, being bound to DNIC for some time. Copper-dependent decomposition of RS-NO causes release of free NO into solution.
S-Nitrosothiol formation at neutral pH. In strongly acidic media RS-NO can be formed with 100% yield in the reaction of thiols with such nyrtosylating agents as nitroso-halogens or higher nitrogen oxides [59, 60]. At neutral pH nitroso-halogens and higher nitrogen oxides are easily hydrolyzed with the formation of nitrite anion. Under these conditions synthesis of RS-NO is sharply attenuated. According to Kopenol [72], at pH 7.0 the nitrite anion conversion into nitrosonium ion in the reaction
is characterized by free energy DeltaG of +94 kJ. Since DeltaG = -RT·lnK, the calculation of K = [NO+]/[NO2-] at 37°C gives the value K = 3.7·10-17. This means that at equilibrium conditions at nitrite concentration of 1 mM the concentration of nitrosylating agent NO+ is extremely low, 3.7·10-20 M. Thus, at neutral pH (and equilibrium conditions) thiol S-nitrosylation cannot proceed via this route. Nevertheless, many researches do not leave this mechanism out of consideration. Passing of NO through oxygen-containing strongly buffered GSH solution resulted in S-nitrosylation of this thiol (up to 20%) [73]. It is possible that under these conditions GS-NO formation is coupled to local acidification of the solution by hydrolysis of higher nitrogen oxides which were formed during NO oxidation. This acidification increases the stability of N2O3 and N2O4 thus increasing the possibility of their nitrosylating action on GSH. High stability of forming GS-NO provides its accumulation in the solution at neutral pH. However, this mechanism in the intracellular medium is very doubtful because of the low rate of NO oxidation by oxygen, whose in vivo content in tissues corresponds to the pressure . 20 mm Hg.
We believe that the process of RS-NO formation during NO oxidation to NO+ is more relevant for biological systems. It requires the presence of oxidant possessing redox potential comparable with that of the redox couple NO+/NO (1.21 V [55, 72]). As follows from the previous section, such a process can be realized during NO incorporation into DNIC acting as a source of NO+ for thiol nitrosylation. We already indicated there that this process can proceed in two ways: either thiols bind nitrosonium ion in the coordination sphere of the complex with subsequent release into solution, or S-nitrosylation occurs right after release of nitrosonium ion into solution.
Our experiments revealed that DNIC actually initiated RS-NO formation in aqueous thiol solution at neutral pH, i.e., iron ions as contaminants (1-2 µM) or additions to the solution (not more than 20 µM) catalyze NO conversion into NO+, which was able to bind thiols. Filling a Thunberg vessel (of 100 ml) containing 2 ml of solution of 1-50 mM cysteine or glutathione in 15 mM Hepes-buffer, pH 7-8, with NO under the pressure 50-700 mm Hg in the absence of oxygen followed by 5 min shaking resulted in appearance of pink colored RS-NO detected by absorbance at 340 nm [54, 74]. Similar results were obtained during filling this system with NO and O2 at the molar ratio NO/O2 (air) not less than 40. Pretreatment of these solutions with selective chelator of bivalent iron, 0.25 mM o-phenanthroline, completely inhibited this process in both variants of the experiments (i.e., in strictly anaerobic conditions and in the presence of small quantities of oxygen) [54]. This also blocked DNIC formation. Addition of o-phenanthroline to a solution containing RS-NO did not influence the content of the latter. These results demonstrate that even in the presence of some quantities of air in the vessel which resulted in appearance of higher nitrogen oxides, S-nitrosylation of thiols was realized by iron catalyzing conversion NO into NO+ and not by these oxides [54].
The degree of S-nitrosylation of cysteine or GSH at neutral pH increased during a decrease of thiol concentration. In the absence of air contaminant, the yield in 1-3 mM cysteine or glutathione reached 40% [74]. In the presence of oxygen as contaminant the percent of glutathione incorporation into GS-NO in 1, 5, and 10 mM solution was 100, 80, and 60% [54]. Increase of efficacy of this process during mixing with NO of a small amount of oxygen we would explain by destabilizing influence of higher nitrogen oxides, formed during NO oxidation in this mixture (mainly NO2) on DNIC. This destabilization leads to a shift of equilibrium of DNIC with its components (in Scheme 3) to the right, i.e., to the accumulation of these components including RS-NO. This was especially demonstrated during oxygen addition to NO when the latter contacted with DNIC bound to bovine serum albumin or horse hemoglobin. This procedure led to complete decomposition of this DNIC and formation of RS-NO bound to these proteins. During the subsequent addition of cysteine and Fe2+ to these RS-NO, they decomposed and DNIC with cysteine appeared [54].
The contact of cysteine with NO under anaerobic conditions at pH > 7.5 resulted in cysteine oxidation. S-Nitrosylation of cysteine under these conditions was observed only at pH 7 and below. Addition of Fe2+ (not more than 20 µM) accelerated this process. Mixing of NO with some quantities of oxygen resulted in cys-NO appearance also at alkaline pH. In contrast to cysteine, GSH was nitrosylated under anaerobic conditions and alkaline pH. Addition of Fe2+ in concentration not more than 50 µM increased GS-NO synthesis. At higher iron concentrations (100 µM and higher) the destroying effect of iron on GS-NO appeared to predominate. As a result the steady-state level of this RS-NO determined by its synthesis and breakdown began to decrease [54]. Under similar experiments with NAP, addition of iron (100-150 µM) favored to the synthesis of SNAP. As mentioned earlier, NAP contains the acetyl-acetonate group which can bind iron and therefore protect the RS-NO against its destroying action. In the presence of NO and contaminants of NO2 additions of iron catalyzed only conversion of NO into NO+, i.e., only conversion of SNAP. As the result, all NAP even at concentration 50-100 mM was converted into SNAP [70] (A. F. Vanin et al., unpublished data).
INTERCONVERSION OF RS-NO AND DNIC WITH THIOL-CONTAINING
LIGANDS
So, we considered above that at neutral pH Fe2+ ions can catalyze both synthesis and decomposition of RS-NO as illustrated in Scheme 8.
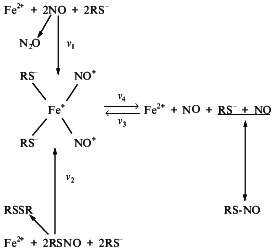
Scheme 8
The predominance of either of these processes is determined by a ratio of Fe2+, NO, thiol, and RS-NO. The central position in these processes belongs to DNIC. In the presence of low-molecular-weight thiols the latter can be formed either in the reaction of NO with Fe2+ (reaction 1) or in the reaction of Fe2+ with RS-NO (reaction 2). If the rate of reaction 1 (v1) is much higher than the rate of DNIC reconstitution from its components (v3) Fe2+ released from DNIC are involved in reaction 1. This results in RS-NO accumulation. This situation occurs under excess NO. At excess RS-NO they are destroyed in the reaction with Fe2+ followed by DNIC formation. If the rate of this reaction (v2) is much higher than v3 (this requires excess of Fe2+ in the system) NO released from DNIC is accumulated.
Thus, the system including NO, thiols, and iron provides coexistence and interconversion of DNIC and RS-NO. Iron plays a special role in this system and determines its appearance. Iron acts as a catalyst which promotes both the conversion of NO into NO+ thereby raising a possibility of thiol S-nitrosylation and decomposition of RS-NO. We found that Fe2+ involved in these processes belongs to a pool of "free" iron (weakly bound in cells and tissues). This iron forms DNIC with thiol-containing ligands which are registered by EPR as a signal 2.03 [41]. Some authors believe that these complexes can be formed as a result of destroying influence of NO on active sites of iron-sulfur proteins [75-78]. This process is suggested to be one manifestation of the cytotoxic effect of NO on animal tissues and cells [79, 80]. However, data concerning the NO effect on iron-sulfur proteins are still contradictory. Initially it was qualitatively demonstrated that NO destroyed active sites of these proteins with DNIC formation [75-77]. Using isolated isoforms of aconitase, interrelationship between deactivation of this iron-sulfur protein in the presence of NO and DNIC formation was demonstrated quantitatively [78]. Nevertheless, NO did not influence the activity of aconitase [81, 82]. Evaluating ratios of iron level in DNIC formed in animal tissues and cells during contact with NO and decrease of iron in the iron-sulfur centers of the respiratory chain during NO action, we did not notice a marked contribution of the "iron-sulfur iron” to DNIC formation [83, 84].
Of course, these incompatible results cannot be considered as incorrect study done by either research group. Many various factors which were not taken into consideration could affected these results. For example, certain buffers, presence of reductants and contaminant metals could influence decomposition iron-sulfur centers in the presence of NO. The presence of iron (especially Fe2+) as contaminant is the most crucial. According to Scheme 2 these ions bind neutral NO molecules and initiate the appearance of NO+ ions in DNIC which are able to nitrosylate thiol ligands or inorganic sulfur in the active sites of iron-sulfur centers, thereby destroying them.
Experiments revealed that nitrosonium ions can actually destroy iron-sulfur centers. Castro et al. [82] found that in contrast to NO, GS-NO rapidly inactivated aconitase probably via S-transnitrosylating action on thiol ligands or inorganic sulfur in the active site of this protein. Using another iron-sulfur protein, pig adrenal adrenodoxin in the isolated pig adrenal tissue we demonstrated a similar effect of GS-NO and SNAP [85]. Treatment of the adrenal tissue with these RS-NO resulted in 3-4-fold decrease of adrenodoxin content evaluated by the intensity of the EPR signal of the reduced form of this protein. The decrease in the level of endogenous substrates is able to reduce this protein potentiated effect. The destroying effect of RS-NO on adrenodoxin was accompanied by marked DNIC formation only in experiments with GS-NO. This suggests that DNIC was not related to decomposition of iron-sulfur centers. In both cases the decomposition was evidently due to the action of nitrosonium ions donated by GS-NO or SNAP.
The ability of DNIC and RS-NO for mutual interconversions suggests possible ways for the change of these NO stabilizing forms which depends on altered levels of iron, low-molecular-weight thiols, and NO in cells and tissues. Decrease in thiol content results in a decrease both of RS-NO and the paramagnetic form of DNIC which is converted into dimeric diamagnetic form. During the decrease of NO concentration at constant level of iron and thiols (usually observed during DNIC and RS-NO transfer from the NO donor cell to the NO accepting cell) the behavior of the DNIC--RS-NO system will vary with the dependence of the availability of iron and thiols. With excess iron RS-NO will disappear faster than DNIC. Model experiments revealed that DNIC can be transformed into mononitrosyl complexes of iron with their subsequent polymerization [86]. With excess thiols the major proportion of bound NO is in the form of RS-NO. Thus, incorporation of NO into DNIC and RS-NO in cells and tissues can lead to the organization of dynamic and, possibly, self-regulating system, where NO can exist either as a free molecule or as included into DNIC and/or RS-NO complexes. The ratio between free NO and its bound forms will be constantly changed. It is possible that EDRF represents such dynamic system.
FACTS AND PROBLEMS
We should emphasize that there are no direct experimental data supporting or refuting the existence in animal cells and tissues of the system of reactions given in Scheme 8. Some indirect data such as physiological properties of DNIC and RS-NO allowing to
compare them with the most studied stabilizing and transport form of NO, EDRF, suggest functioning of this system. RS-NO and DNIC are characterized by high vasodilatation activity comparable in dose ratio (by the amount of incorporated NO) with vasodilatation action of EDRF [12, 17, 44, 58, 87-89]. However, the comparison of the duration of vasodilatation effects of these compounds requires further investigation. Feelisch et al. [90] demonstrated that the pass of a drop of medium containing EDRF or cys-NO between three cascade-disposed dilating rings of rabbit aorta was accompanied by an equal attenuation of vasodilating effect of both compounds. This suggests similar life-time of EDRF and cys-NO. However, cysteine addition to the medium sharply increased the duration of the vasodilating effect of cys-NO. The authors interpreted this result in terms of binding by cysteine of contaminant metals destroying cys-NO, and it was concluded that EDRF and this RS-NO are different molecules. The same conclusion was made in the case of DNIC. The life-time of DNIC as a vasodilator was incomparably longer than that of EDRF: efficacy of vasodilating effect of DNIC remained unchanged over a drop passing in the cascade system. Free NO behaved as EDRF in this system.
The major argument against the conclusion of this study rejecting cys-NO or DNIC as candidates for EDRF consists of the fact that cys-NO and DNIC were administered into the cascade system of vascular preparations in medium which differed from the medium used for EDRF administration. Besides EDRF, other agents (e.g., NO2) could pass from the endothelial cell culture or a vessel with intact endothelium into the incubation medium. We found that NO2 can sharply attenuate DNIC stability and due to the interrelationship with RS-NO, influence its stability as well. The influence of NO2 on vasodilating activity of DNIC and RS-NO could be checked by co-administration of NO in the cascade system. Its oxidation to NO2 can accelerate DNIC decomposition and alter behavior of S-nitrosothiols. The experimental verification of possible influence of NO2 and other agents secreted by endothelial cells in response to the action of biologically active compounds stimulating EDRF synthesis in them seems to be very important for the comparison of physiological activity of EDRF and the system DNIC--RS-NO--NO--thiols discussed in the present review.
In this system iron acts as the main component determining NO incorporation into DNIC or RS-NO. If this system is related to EDRF, iron chelators must influence the life-time and efficacy of the vasodilating effect of EDRF. We have started such studies [89] and obtained results that at least do not contradict the hypothesis of EDRF nature considered here. Administration of chelator of transition metal ions diethyldithiocarbamate (DETC) to isolated rat vessels pre-dilated by acetylcholine (via EDRF) or DNIC caused vasoconstriction. We suggest that as in the case of treatment with DNIC in vessels dilated by EDRF, iron-containing store of NO (probably DNIC) appears. This maintains vessels in the relaxed state. Decomposition of this store by DETC restores vascular tone. Vasodilatation was less effective when acetylcholine or DNIC were added to the system pretreated with DETC. However, the residual dilatation was abolished neither by hemoglobin (as NO acceptor) nor by DETC. It is suggested that both ways initiating vasodilatation in vascular tissue by DETC resulted in appearance of some compound capable of maintaining vasodilatation for a long time. In view of the cGMP-independent vasodilating effect of NO, due to its influence on ion channel conductivity [91, 92], it is possible that this effect of DETC is related to changes in the functioning of these channels. In the experiments with DNIC, nitrosonium ions released from this complex by DETC could cause such changes.
Another problem is also important for elucidation of the role of DNIC in the functioning of NO in animals and humans. The formation of these complexes in animal organs in vivo during initiation of increased NO synthesis from L-arginine by, e.g., bacterial endotoxin, has not yet been demonstrated. Under these conditions DNIC formation is easily detected in various animal cell cultures after synthesis of inducible NOS (5-6 h after cell contact with an activator of the synthesis of this enzyme) [22-26]. In the whole body DNIC formation was detected only a few days after the systemic consumption with drinking water of sodium nitrite as an exogenous source of NO [36-40] or a few days after the infection of animals with pathogenic bacteria initiating synthesis of inducible NOS [93]. It is possible that, as in cell cultures, DNIC appeared in animal organs simultaneously with stimulation of NO synthesis attributed to the appearance of inducible NOS in them. However, due to rapid decomposition, the steady-state level of these complexes is too low to be measured by the EPR method. The intensive decomposition of DNIC is due to the destabilizing action of higher nitrogen oxides formed from NO, and this results in the accumulation of only RS-NO described in Scheme 8. Long-term production of NO in the body causes accumulation of reductants decreasing the level of higher nitrogen oxides, and this leads to DNIC accumulation in organs. This hypothesis is currently investigated by our group.
The possible penetration through the cell membrane of DNIC and RS-NO requires detailed investigation. Only indirect data support this possibility. We found that the administration to mice of DNIC with thiol ligands containing 57Fe resulted in the appearance of DNIC-bound proteins in liver cells. This conclusion was based on the conservation of 57Fe in these complexes and the specific shape of the EPR signal typical for DNIC located inside liver cells [38, 39, 94, 95]. However, the mechanism of DNIC penetration into liver cells remains unclear: whether DNIC components penetrate separately with subsequent reconstitution inside cells or DNICs pass cell membrane as the whole molecule. It should be noted that one-electron reduction of these complexes providing neutralization of their total charge might promote DNIC penetration through the cell membrane [52]. Similar questions arise during consideration of RS-NO penetration into cells. S-Nitrosylation of hemoglobin during contact of low-molecular-weight RS-NO with erythrocytes seems to support such possibility [96]. However, it remains unclear how these compounds do appear inside cells. Do they penetrate cell membrane as the whole molecule or they are initially converted into DNICs, which pass into cells and possibly S-nitrosylate hemoglobin? These questions require further investigations.
The other interesting question consists of the influence of NO incorporation into RS-NO or DNIC on the cytotoxic effect of NO due to its conversion into peroxynitrite (ONOO-) [97]. Incorporation of NO into these compounds can obviously decrease the possibility of such conversion and therefore attenuate its cytotoxic action. Only free NO is able to react with superoxide anions with peroxynitrite formation. Thus, formation of DNIC and RS-NO can be considered as factors regulating the effectiveness of cytotoxic action of NO provided that this action is mainly determined by peroxynitrite.
This work was supported by the Russian Foundation for Basic Research (grant No. 96-04-48066).
REFERENCES
1. Moncada, S., Palmer, R. M. J., and Higgs, E. A.
(1991) Pharmacol. Rev., 43, 109-142.
2. Stamler, J. S., Singel, D. J., and Loscalzo, J.
(1992) Science, 258, 1898-1902.
3. Nathan, C. (1992) FASEB J., 6,
3051-3064.
4. Dawson, T. M., and Snyder, S. H. (1994) J.
Neurosci., 14, 5147-5159.
5. Brüne, B., Dimmeler, S., Molina y Vedia, L.,
and Lapetina, E. G. (1994) Life Sci., 54, 61-70.
6. Iyengar, R., Stuehr, D. J., and Marletta, M. A.
(1987) Proc. Natl. Acad. Sci. USA, 84,
6369-6373.
7. Hibbs, J. B., Jr., Taintor, R. R., Vavrin, Z., and
Rachlin, E. M. (1989) Biochem. Biophys. Res. Commun.,
157, 87-94.
8. Clancy, R. M., and Abramson, S. B. (1995) Proc.
Soc. Exp. Biol. Med., 210, 93-100.
9. Lancaster, J. R. (1994) Proc. Natl. Acad. Sci.
USA, 91, 8137-8141.
10. Furchgott, R. F., and Zawadzki, J. V. (1980)
Nature, 288, 373-376.
11. Palmer, R. M. J., Ferrige, A. G., and Moncada,
S. (1987) Nature, 327, 524-526.
12. Myers, P. R., Minor, R. L., Guerra, R., Bates,
J. N., and Harrison, D. G. (1990) Nature, 345,
161-163.
13. Myers, P. R., Guerra, R., and Harrison, D. G.
(1992) J. Cardiovasc. Pharmacol., 20, 392-400.
14. Rubanyi, G. M., Johns, A., Wilcox, D., Bates, J.
N., and Harrison, D. G. (1991) J. Cardiovasc. Pharmacol.,
17 (Suppl. 3), S41-S45.
15. Vedernikov, Y. P., Mordvintcev, P. I., Malenkova, I. V., and
Vanin, A. F. (1990) in Nitric Oxide from L-Arginine: a Bioregulatory
System (Moncada, S., and Higgs, E. A., eds.) Elsevier Science
Publishers B. V., Amsterdam, pp. 373-377.
16. Vanin, A. F. (1991) FEBS Lett.,
289, 1-3.
17. Vedernikov, Y. P., Mordvintcev, P. I.,
Malenkova, I. V., and Vanin, A. F. (1992) Eur. J. Pharmacol.,
211, 313-317.
18. Stamler, J. S., Jaraki, O., Osborne, J., Simon,
D. I., Keaney, J., Vina, J., Singel, D., Valery, C. R., and Loscalzo,
J. (1992) Proc. Natl. Acad. Sci. USA, 89,
7674-7677.
19. Gaston, B., Reilly, J., Drazen, J. M., Fackler,
J., Ramdev, P., Arnelle, D., Mullins, M. E., Sugarbaker, D. J., Chee,
C., Singel, D. J., Loscalzo, J., and Stamler, J. S. (1993) Proc.
Natl. Acad. Sci. USA, 90, 10957-10961.
20. Stamler, J. S., Osborne, J. A., Jaraki, O.,
Rabbani, L. E., Mullins, M., Singel, D., and Loscalzo, J. (1993) J.
Clin. Invest., 91, 308-318.
21. Clancy, R. M., Levartovsky, D.,
Leszczynska-Piziak, J., Yegudin, J., and Abramson, S. B. (1994)
Proc. Natl. Acad. Sci. USA, 91, 3680-3684.
22. Lancaster, J. R., and Hibbs, J. B. (1990)
Proc. Natl. Acad. Sci. USA, 87, 1223-1227.
23. Drapier, J.-C., Pellat, C., and Henry, Y. (1991)
J. Biol. Chem., 266, 10162-10167.
24. Stadler, J., Bergonia, H. A., DiSilvio, M.,
Sweetland, M. A., Billiar, T. R., Simmons, R. L., and Lancaster, J. R.
(1993) Arch. Biochem. Biophys., 302, 4-11.
25. Mülsch, A., Mordvintcev, P. I., Vanin, A.
F., and Busse, R. (1993) Biochem. Biophys. Res. Commun.,
196, 1303-1308.
26. Geng, Y.-L., Petersson, A.-S., Wennmalm, A., and
Hannson, G. (1994) Exp. Cell Res., 214, 418-424.
27. Vanin, A. F., and Nalbandyan, R. M. (1965)
Biofizika, 10, 167-168.
28. Vithaytil, A. J., Ternberg, J. L., and Commoner,
B. (1965) Nature, 207, 1246-1249.
29. Vanin, A. F., Blumenfeld, L. A., and
Chetverikov, A. G. (1967) Biofizika, 12, 829-841.
30. Vanin, A. F. (1967) Biokhimiya,
32, 228-232.
31. Woolum, J. C., Tiezzi, E., and Commoner, B.
(1968) Biochim. Biophys. Acta, 160, 311-320.
32. Vanin, A. F., and Blumenfeld, L. A. (1969) in
Abstract Book of Int. Jubilee Conf. on EPR, Kazan Universiry
Press, Kazan, p. 25.
33. Woolum, J. C., and Commoner, B. (1970)
Biochim. Biophys. Acta, 201, 131-140.
34. Vanin, A. F., Kubrina, L. N., Lisovskaya, I. L.,
Malenkova, A. V., and Chetverikov, A. G. (1971) Biofizika,
16, 650-658.
35. Vanin, A. F., and Chetverikov, A. G. (1968)
Biofizika, 13, 608-613.
36. Foster, M. A., and Hutchison, J. M. S. (1974)
Phys. Med. Biol., 19, 289-302.
37. Vanin, A. F., Kiladze, S. V., and Kubrina, L. N.
(1977) Biofizika, 22, 850-857.
38. Vanin, A. F., Kiladze, S. V., and Kubrina, L. N.
(1978) Biofizika, 23, 474-479.
39. Vanin, A. F., and Varich, V. Ya. (1981) Stud.
Biophys., 86, 175-185.
40. Varich, V. Ya., and Vanin, A. F. (1983)
Biofizika, 28, 1055-1060.
41. Vanin, A. F. (1980) Nitrosyl Complexes of
Non-heme Iron in Animal Tissues and Microorganisms: Doctoral
dissertation[in Russian], Institute of Chemical Physics, Moscow.
42. McDonald, C. C., Phillips, W. D., and Mower, H.
F. (1965) J. Am. Chem. Soc., 87, 3319-3326.
43. Vanin, A. F., Malenkova, I. V., Mordvintcev, P.
I., and Mülsh, A. (1992) Biokhimya, 58,
1094-1103.
44. Vanin, A. F., Stukan, R. A., and Manukhina, E.
B. (1996) Biochim. Biophys. Acta, 1295, 5-12.
45. Jocelyn, P. C. (1972) Biochemistry of SH
Group, University Edinburgh.
46. Burbaev, D. Sh., Vanin, A. F., and Blumenfeld,
L. A. (1971) Zh. Strukt. Khim., 12, 252-256.
47. Martini, G., and Tiezzi, E. (1971) Trans.
Faraday Soc., 67, 2538-2547.
48. Butler, A. R., Glidewell, C., and Li, M.-H.
(1988) Adv. Inorg. Chem., 32, 335-393.
49. Bryar, T. R., and Eaton, D. R. (1992) Can. J.
Chem., 70, 1917-1926.
50. Thomas, J. T., Robertson, J. H., and Cox, E. G.
(1958) Acta Crystallogr., 11, 599-607.
51. Baltusis, L. M., Karlin, K. D., Rabinowitz, H.
N., Dewan, J. C., and Lippard, S. J. (1980) Inorg. Chem.,
19, 2627-2632.
52. Burbaev, D. Sh., and Vanin, A. F. (1973)
Dokl. Akad. Nauk SSSR, 213, 860-863.
53. Pearsall, K. A., and Bonner, F. T. (1982)
Inorg. Chem., 21, 1978-1985.
54. Vanin, A. F., Malenkova, I. V., and Serezhenkov,
V. A. (1997) Nitric Oxide: Biol. Chem., 1, 191-203.
55. Henry, Y. A., Guissani, A., and Ducastel, B.
(1997) Nitric Oxide Research from Chemistry to Biology: EPR
Spectroscopy of Nitrosylated Compounds, R. G. Landes Company,
Austin, Texas, USA, p. 18.
56. Schmidt, H. H. H. W., Hofmann, H., Schindler,
U., Shutenko, Z. S., Cunningam, D. D., and Feelisch, M. (1996) Proc.
Natl. Acad. Sci. USA, 93, 14492-14497.
57. Xia, Y., and Zweier, J. L. (1997) Proc. Natl.
Acad. Sci. USA, 94, 12705-12710.
58. Mülsch, A., Mordvintcev, P. I., Vanin, A.
F., and Busse, R. (1991) FEBS Lett., 294,
252-256.
59. Oae, S., and Shinhama, K. (1983) Org. Prep.
Proc. Int., 15, 165-198.
60. Butler, A. R., Flitney, F. W., and Williams, D.
L. H. (1995) Trends Physiol. Sci., 16, 18-23.
61. Mathews, W. R., and Kerr, S. W. (1993) J.
Pharmacol. Exp. Ther., 267, 1529-1537.
62. Feelisch, M., and Stamler, J. S. (1996) in
Methods in Nitric Oxide Research (Feelisch, M., and Stamler, J.
S., eds.) John Willey and Sons Ltd, New York, p. 84.
63. Saville, B. (1958) Analyst, 83,
670-672.
64. Forth, W., and Rummel, W. (1973) Physiol.
Rev., 53, 724-732.
65. Vanin, A. F. (1995) Biochemistry
(Moscow), 60, 593-601 (Russ.)
66. Sammes, P. G., and Yahioglu, G. (1994) Chem.
Soc. Rev., No. 2, 327-334.
67. Askew, S. C., Butler, A. R., Flitney, F. W.,
Kemp, G. D., and Megson, I. L. (1995) Bioorg. Med. Chem.,
3, 1-9.
68. Gorren, A. C. F., Schrammel, A., Schmidt, K.,
and Mayer, B. (1996) Arch. Biochem. Biophys., 330,
219-228.
69. Dicks, A. P., Swift, H. R., Williams, D. L. H.,
Butler, A. R., Al-Sa`doni, H. H., and Cox, B. G. (1996) J. Chem.
Soc. Perkin Trans., 2, 481-487.
70. Vanin, A. F., Malenkova, I. V., and Serezhenkov,
V. A. (1998) Proc. V Int. Meet. Biology of Nitric Oxide (Kyoto),
Japan Pharmacol. Soc., Kyoto, in press.
71. Vanin, A. F. (1995) Biochemistry
(Moscow), 60, 225-229 (Russ.).
72. Kopenol, W. H. (1996) Meth. Enzymol. Pt.
A, 268, 7-12.
73.Kharitonov, V. G., Sundquist, A. R., and Sharma,
V. S. (1995) J. Biol. Chem., 270, 28158-28164.
74. Vanin, A. F., and Malenkova, I. V. (1996)
Biochemistry (Moscow), 61, 374-379 (Russ).
75. Reddy, D., and Lancaster, J. R. (1983)
Science, 221, 769-770.
76. Stuehr, D. J., and Nathan, C. F. (1989) J.
Exp. Med., 169, 1543-1555.
77. Welter, R., Yu, L., and Yu, C.-A. (1996)
Arch. Biochem. Biophys., 331, 9-14.
78. Kennedy, M. C., Antholine, W. E., and Beinert,
H. (1997) J. Biol. Chem., 272, 20340-20347.
79. Drapier, J.-C., and Hibbs, J. B. (1988) J.
Immunol., 140, 2829-2838.
80. Hibbs, J. B., Taintor, R. R., Vavrin, Z., and
Rachlin, E. M. (1988) Biochem. Biophys. Res. Commun.,
157, 87-94.
81. Hausladen, A., and Fridovich, I. (1994) J.
Biol. Chem., 269, 29405-29408.
82. Castro, L., Rodriguez, M., and Radi, R. (1994)
J. Biol. Chem., 269, 29409-29415.
83. Vanin, A. F. (1987) Biofizika, 31,
128-132.
84. Vanin, A. F., Men'shikov, G. B., Moroz, I. A.,
Mordvintcev, P. I., Serezhenkov, V. A., and Burbaev, D. Sh. (1992)
Biochim. Biophys. Acta, 1135, 275-279.
85. Voevodskaya, N. V., Khrapova, N. V., and Vanin,
A. F. (1997) in Abst. Int. Symp. Nitric Oxide in Health and
Diseases (Kumamoto), University Kumamoto, p. 15.
86. Burbaev, D. Sh., and Vanin, A. F. (1970)
Dokl. Akad. Nauk SSSR, 190, 1348-1351.
87. Kowaluk, E. A., and Fung, H.-L. (1990) J.
Pharmacol. Exp. Ther., 255, 1256-1264.
88. Zamora, R., and Feelisch, M. (1994) Biochem.
Biophys. Res. Commun., 201, 54-62.
89. Vedernikov, Y. P., Mordvintcev, P. I.,
Malenkova, I. V., and Vanin, A. F. (1992) Eur. J. Pharmacol.,
212, 125-128.
90. Feelisch, M., te Poel, M., Zamora, R., Deussen,
A., and Moncada, S. (1994) Nature, 368, 62-65.
91. Bolotina, V. M., Najibi, S., Palacino, J. J.,
Pagano, P. J., and Cohen, R. A. (1994) Nature, 368,
850-853.
92. Taguchi, H., Heistad, D. D., Chu, Y., Rios, D.,
Ooboshi, H., and Faraci, F. M. (1996) J. Pharmacol. Exp. Ther.,
279, 1514-1519.
93. Chamulitrat, W., Jordan, S. I., Mason, R. P.,
Litton, A. L., Wilson, J. G., Wood, E. R., Wolberg, G., and Molina y
Vedia, L. (1995) Arch. Biochem. Biophys., 316,
30-37.
94. Vanin, A. F., Kubrina, L. N., and Aliev, D. I.
(1980) Stud. Biophys., 80, 221-230.
95. Vanin, A. F., Kurbanov, I. S., Mordvintcev, P.
I., and Aliev, D. I. (1987) Stud. Biophys., 120,
145-154.
96. Jia, L., Bonaventura, C., Bonaventura, J., and
Stamler, J. S. (1996) Nature, 380, 221-226.
97. Beckman, J. S., and Kopenol, W. (1996) Am. J.
Physiol., 271, C1424-C1437.