REVIEW: The Role of Nitric Oxide Chemistry in Cancer Treatment
D. A. Wink1*, Y. Vodovotz2, J. A. Cook1, M. C. Krishna1, S. Kim1, D. Coffin1, W. DeGraff1, A. M. Deluca1, J. Liebmann1, and J. B. Mitchell1
1Radiation Biology Branch, National Cancer Institute, Bldg. 10, Room B3-B69, Bethesda, MD 20892, USA; fax: (301) 480-2238; E-mail: wink@box-w.nih.gov2Cardiology Research Foundation and Medlantic Research Institute, Washington, DC 20010, USA
* To whom correspondence should be addressed.
Received January 28, 1998
Over the last decade the role of nitric oxide (NO) in various disease states has become apparent. In cancer, NO plays a variety of roles which are at times contradictory. On one hand, NO is involved in different etiological mechanisms as well as promoting tumor growth. Yet, NO derived from leukocytes plays a seminal role in their tumoricidal activity. In cancer treatment, NO also has diverse effects. Whereas in vitro, NO can enhance the cytotoxic efficacy of some chemotherapeutic agents as well as radiation, NO donors can provide whole body protection against these same agents. This manuscript will discuss some mechanisms involved with NO and cancer treatment modalities and the potential application of these findings to cancer therapy.
KEY WORDS: nitric oxide, cancer, oncology
The treatment of cancer has been one of man's most intensive and greatest challenges. As our knowledge has increased in biology over the century the resultant basic scientific discoveries have led to potential strategies for more effective treatments for cancer. Over the last decade a new molecule, nitric oxide (NO), has emerged as the one the most active areas of concentration in biomedical research. This molecule has been shown to regulate numerous physiological functions as well as participating in various pathological conditions.
The participation of NO in the tumoricidal activity of macrophages was one the first roles reported for this molecule [1]. Inhibition of NO produced by macrophages lowered the antitumor response, and this seminal finding was one of the major contributions to the explosion of research focused on the function and control of NO in normal and disease states. Though NO may be an important factor in host defense against cancer, other reports suggest that NO promotes tumor growth [2, 3]. It appears that this dichotomy depends largely on what stage of cancer NO is present, as well as on the quantities of NO produced. Thus, devising strategies to utilize modification of NO requires an understanding of factors such as timing, concentration, and location of this radical (Table 1). In this report, we will discuss how NO relates to cancer biology, as well as potential strategies to improve clinical cancer treatment modalities via modulation of NO.
Table 1. Properties of NO in cancer
biology
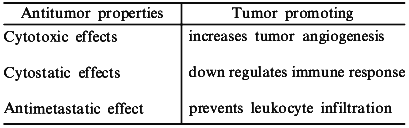
In order to understand the diverse effects of NO in cancer and cancer treatment, it is useful to discuss some the basic chemical and biochemical properties of this diatomic radical. Nitric oxide is derived in vivo from the enzyme nitric oxide synthase [4-8] (Fig. 1). There are three isoforms of this enzyme: type I (neuronal nitric oxide synthase, nNOS; NOS-1), type II (inducible nitric oxide synthase, iNOS; NOS-2), and type III (endothelial nitric oxide synthase, eNOS; NOS-3). Type I and III are usually constitutively present in the cell (cNOS); however, under certain conditions, their expression can also be induced. These isoenzymes are activated by an increase in intracellular calcium, which facilitates the binding of calmodulin to NOS, thus activating the enzyme [7]. The second class of NOS, the so-called "inducible" form, is expressed in cells after exposure to certain cytokines; however, some tissues express this isoform of NOS even under basal conditions. The major difference between the “constitutive” and “inducible” isoforms is the amount and duration of NO produced by either NOS. All three isoforms have similar specific activities when purified to homogeneity; however, the total amount of NO generated per cell by cNOS is low as compared to that generated by iNOS. The flux of NO generated by cNOS is of short duration, while iNOS generates considerably higher concentrations of NO for periods of hours to days. Therefore, physiological versus potentially toxic actions of NO might be dictated by the presence and activity of specific isoforms of NOS. As we have previously described, the difference in fluxes of NO is critical to the understanding of the chemical reactions involving NO in vivo and the ultimate biological effect.
The concept of the “chemical biology of NO” takes into account the reactions involving this radical which can occur in vivo, and discusses them in the context of relevant biological effects [9, 10]. Conceptually, the chemical biology of NO provides a guide to the location and timing of the chemical reactions that NO can undergo in vivo. The chemical biology of NO encompasses two distinct categories, consisting of direct and indirect effects (Fig. 1). Direct effects are defined as those chemical reactions in which NO will interact directly with a biological target. Indirect effects are defined as the chemistry mediated by reactive nitrogen oxide species (RNOS) derived from the interaction of NO with superoxide (O2-) or oxygen (O2). A variety of RNOS can be formed by these interactions, leading to specific types of chemistry such as nitrosation, oxidation, nitration, and hydroxylation. The predominating chemistry by the array of metabolites that are actually detectable in vivo appear to be nitrosation and oxidation. These two types of indirect effects can exert different effects on biological systems (Fig. 1). By understanding the sources of these reactions, it will be easier to evaluate the relevance of a particular reaction for the biological effect of NO in the target tissue. This would allow the evaluation of what role NO has in cancer and potential treatment strategies.Fig. 1. The source of NO and the chemical effects in biology.
ROLE OF NO IN CANCER
In order to understand the potential uses of NO, it is necessary to discuss some of the known roles for this molecule in cancer biology. Macrophages were first shown to secrete NO causing respiratory distress in lymphoma cells [1, 11]. Further reports demonstrated that large amounts of NO derived from iNOS expressed in other cell types such as Kupffer cells, natural killer cells (NK), microglial and endothelial cells participates in the tumoricidal activity against many types of tumors [12-17]. The combined findings of these cell culture experiments imply that NO has a cytostatic or cytotoxic effect on tumor cells.
In vivo studies suggest that NO plays an important role in the control of tumor growth. Macrophages harvested from tumor-bearing animals exhibit reduced capacity to produce NO as well as diminished tumoricidal activity [18-20]. Reduction in the expression or activity of iNOS is thought to be due in part to systemic formation of tumor-derived suppressor agents such as interleukin-10 (IL-10), tumor growth factor beta1 (TGF-beta1), and prostaglandin E2 (PGE2) [21, 22] which reduce both NO production and tumoricidal activity by macrophages. Studies have shown that tumor-bearing patients have elevated levels of IL-10 and TGF-beta1 [21-23] supporting the notion that there is a relationship between these suppressive factors, reduction of NO production, and tumor burden. In addition to these agents, other agents such as phosphatidylserine secreted from mammary tumors also decrease NOS activity in leukocytes [24]. The relationship between reduction of NO and increased tumor burden indicates that the presence of this free radical is an integral part of the antitumor response of the immune system.
Despite the cytotoxic and cytostatic properties of NO in the tumoricidal activity of the immune system, studies have indicated that NO can be an important mediator of tumor growth. Various tumors possess both constitutive and inducible forms of NOS, which implies that NO may be an important factor in different stages of tumor progression. Human breast tumors [3], cervical tumors [25], tumors associated with the central nervous system [26], colon [2], and head and neck [27] cancers all have been shown to have elevated levels of NOS. The presence of NOS suggest that NO produced by these cells is part of the basic mechanisms which promote tumor growth.
A comparison of different tumor cell lines can illustrate how endogenous NO can have very different influences on tumor growth. Fidler and coworkers examined the effect of endogenous production of NO on the growth of melanoma cellsboth in cell culture and in whole animals [28, 29]. These authors reported that melanoma cells transfected with the iNOS gene or stimulated with pro-inflammatory cytokines to induce expression of iNOS exhibit reduced cell growth in vitro as well as limited tumorigenesis and metastasis in vivo. Other tumor cells expressing iNOS show very different results, however. Human adenocarcinoma (DLD-1) and murine mammary carcinoma (EMT-6) expressing iNOS were growth-inhibited in vitro, similar to melanoma cells. However, when either cell line was transplanted into mice, they became considerably more aggressive than there counterparts which did not express iNOS [2, 30]. Other studies suggest that the activity of the chemotherapeutic drug 5-fluorodeoxyuridine (5-FUdR) in colon cancer may be in part due to reduction of iNOS expression [31]. Furthermore, biopsies of human mammary tumors demonstrated a greater expression of iNOS in tumors of higher grades, which are more invasive [2].
Other studies indicate that NO augments tumor growth by modulating processes involved in tumor physiology. For instance, NO influences production of prostaglandin (PGE2) synthesis. Several studies suggest that NO can shift the balance in arachidonic acid metabolism to favor prostaglandin synthase while limiting lipoxygenase products [32-39]. Increased production of PGE2 can enhance capillary leakiness in tumors [40]. Elevated levels of PGE2 cause capillaries to increase the passage of small molecules into tissue. Several studies have shown that tumor vascular permeability is mediated by NO produced in tumor cells themselves [41, 42]. In addition to increasing nutrient levels, PGE2 is a tumor suppressive factor which suppresses macrophage tumoricidal activity [43]. Therefore, enhancement of PGE2 by NO originating from the tumor cell may suppress leukocyte-derived NO production and concomitant tumoricidal activity of macrophages, while keeping the tumor vasculature open to nutrients and thus allowing rapid growth.
Several studies show that T-cell proliferation is dramatically reduced by NO, and this effect may also underlie some of the negative effects of NO in cancer. Administration of NO inhibitors also resulted in increased activity of lymphokine-activated killer cells, which reduced tumorigenesis [42]. This result may indicate that NO is essential in controlling T-cell proliferation at the tumor as well at remote sites. Therefore, NO may influence a number of factors which promote tumor growth, both positively and negatively.
The use of NOS inhibitors may have antitumor properties which might: 1) decrease capillary leakiness; 2) increase leukocyte infiltration; 3) increase systemic macrophage activity by elevating lipoxygenase levels and decreasing prostaglandins; and 4) increase T-cell proliferation. A recent study also shows that tissue leakiness mediated by IL-2 therapy is abated by NOS inhibitors, thus preventing unwanted side effects of treatment with this cytokine [42]. Though NO at the cellular level can mediate tumoricidal activity, there are clearly more complex effects of NO in vivo which have to be considered carefully. These results suggest that though NO may have cytostatic and cytotoxic properties; inhibition of NO production at specific stages of tumor development also may be beneficial.
USE OF NO IN EXISTING MODALITIES OF CANCER TREATMENT
As discussed above, NO can cause cell death in some tumor cells such as those of leukocyte origin. However, studies in which other tumors cells were treated with NO via NO donor complexes in vitro revealed that this diatomic molecule is not an effective tumoricidal agent [44-47]. These studies suggest that NO donors themselves may not be useful anti-neoplastic agents. However, recent studies have suggested that NO donors can enhance some clinically important modalities of cancer treatment [45, 46]. In this section, we will discuss some of the mechanisms by which NO donor compounds may improve the efficacy of both radiotherapy and chemotherapy.
Radiotherapy. One of the major factors that limits the effectiveness of radiation therapy is the presence of radioresistant hypoxic tumor cell populations. Over the past 50 years, there has been a concerted effort to identify agents which are effective hypoxic cell radiosensitizers. To date, there have been few substances which are able to effectively overcome the hypoxic effect. However, several papers have reported that NO can radiosensitize hypoxic cells extensively. Howard-Flanders first showed in 1957 that NO radiosensitized hypoxic bacteria [48]. Nearly four decades later, it was shown that NO and NO donor compounds effectively radiosensitize hypoxic mammalian cells too, with a substance enhancement ratio (SER) of 2.4 [49]. These findings demonstrate that NO is equally as effective as oxygen at enhancing the sensitivity of hypoxic cells to radiation.
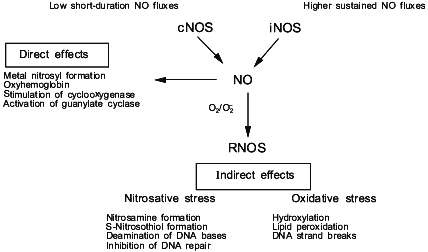
NO-mediated radiosensitization of hypoxic cells is proposed to occur via a mechanism similar to that of O2 [48, 49]. Carbon-centered radicals are initially generated by ionizing radiation on DNA. In the absence of NO or O2, these reactive radicals scavenge nearby protein hydrogen atoms, facilitating DNA repair (Fig. 2). This process limits the number of DNA lesions per photon. However, it has been postulated that NO reacts with these high energy carbon-centered radicals to form complexes which are not capable of abstracting protein hydrogen atoms. The fixing of the radiation induced damage increases the number of lesions per photon, hence increasing radiation-mediated cell death. An advantage of using NO as an hypoxic cell radiosensitizer is that it penetrates into tissue farther than oxygen due to its higher diffusion coefficient. The lower concentrations of NO and higher diffusion rate suggest that NO may be an ideal candidate for new strategies in radiotherapy.
An evaluation of various NO donor complexes reveal that not all of these compounds are capable of radiosensitization of hypoxic cells. SIN-1 and sodium nitroprusside (SNP), which are NO donors used clinically to control vascular tone, did not sensitize hypoxic cells to radiation appreciably [47]. However, the S-nitrosothiols, S-nitrosoglutathione (GSNO) and S-nitroso-N-acetylpenicillamine (SNAP), were excellent hypoxic cell radiosensitizers [47]. In fact, SNAP, was 10 times more potent than the NONOate DEA/NO (diethylamine--NO complex) (Table 2). These findings may indicate that S-nitrosothiols may prove to be one of the most useful compounds for increasing the radiosensitization of hypoxic cells in tumors.Fig. 2. Mechanism for radiosensitization of hypoxic cells by NO and O2.
Table 2. Substance enhancement ratio (SER)
of NO donor agents (from [47, 50])
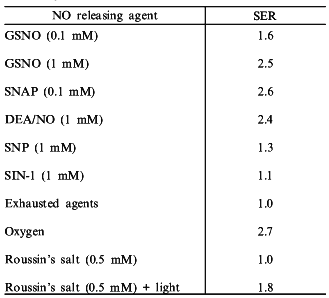
In addition to exploring the use of compounds which release NO under physiological conditions, photochemical methods for the delivery of NO to hypoxic cells has been explored. Roussin's salt is an iron--sulfur complex containing four NO ligands which upon photolysis releases NO [50]. Roussin's salt is part of class of iron sulfur complexes which have been detected in tumor cells in the presence of activated macrophages and may provide insights into potential strategies by which to deliver NO site-specifically [51-53]. In the absence of light (>520 nm), no radiosensitization was observed. However, simultaneous irradiation and exposure of hypoxic cells to light, which released NO from Roussin's salt, resulted in significant enhancement of radiosensitization (Table 2) [50]. Electrochemical methods verified that NO was indeed produced through photolysis. These experiments demonstrate that compounds which release NO either thermally or photochemically can be effective hypoxic cell radiosensitizers.
Though NO can sensitize cells to radiation, NO can influence physiological functions which may alter the radiation response. One of the principal effects of NO is the reduction of systemic blood pressure. Agents which modulate vascular tone can provide protection against whole body radiation in animals [54]. NOS inhibitors and NO donors were evaluated as protective agents against whole body irradiation. Administration of NG-nitro-L-arginine between 15 and 60 min before irradiation resulted in protection against whole body radiation. The LD50/30 (the dose required to kill 50% of the mice after 30 days) was 10.67 Gy with the inhibitor present while the LD50/30 in the absence of the inhibitor was 8.22 Gy. Contrary to the cell culture experiments, administration of DEA/NO 10 or 30 min before irradiation resulted in marked protection [54]. The LD50/30 was 10.63 Gy for animals treated 10 min with the NO donor and 9.46 Gy for animals treated 30 min prior to radiation.
Similar protective effects of both these agents were unexpected since one delivers NO, causing hypotension, and the other inhibits NO production, causing hypertension. Upon closer examination, however, this discrepancy is not as unexpected as it first may seem. The mechanism by which NG-nitro-L-arginine protects is readily apparent. At the doses of radiation used in this study, radiation induced bone marrow damage is the cause of death. Administration of NG-nitro-L-arginine increases blood pressure which results in reduced blood flow to the bone marrow. This creates hypoxia in this region. Hence the generation of hypoxia protects the cells in the bone marrow from radiation. A marker of hypoxic tissue [14C]etanidazole was used to determine the extent of hypoxia induced by NG-nitro-L-arginine. There was marked increase of this marker of hypoxia in bone marrow in animals treated with NG-nitro-L-arginine [54].
The mechanism for protection by DEA/NO is less obvious. DEA/NO causes hypotension which can decrease blood flow to bone marrow and gut via the vascular "steel" effect. This reduction then brings about hypoxia in the bone marrow and gut regions which protects these tissues from radiation damage [54]. As with the NOS inhibitors, the presence of [14C]etanidazole was increased in the bone marrow, which supports the contention that hypoxia was induced in this tissue.
Can modulation of NO be protective of normal tissue while increasing efficacy of radiotherapy for tumors? The NO donor SIN-1 showed an increase in ATP production in tumors exposed to radiation, results which suggest a modest increase in radiosensitization [55]. However, cell culture experiments showed that SIN-1 did not sensitize either aerobic or hypoxic cells to radiation, which implies that these effects may be due to other mechanisms [47]. It has been suggested that tumor blood flow is increased following treatment with NO donors, due to changes in tumor vascular tone [54]. It has been shown that hypotension induced by hydralazine increases tumor hypoxia [56]. This was due to the vascular steel effect, which shunted blood flow from the tumor to other tissues.
We have started to explore the effect of NO donors on radiotherapy of tumors in vivo. Preliminary results using DEA/NO in conjunction with radiation show mixed results. In experiments performed in vivo, irradiated squamous cell carcinoma (SCC) tumor cells exhibited increased tumor regrowth delay in the presence of DEA/NO. This finding suggests that this NO donor may improve the efficacy of ionizing radiation in this model. These findings are consistent with the work of Wood et al. [55], who suggested that NO may improve radiation treatment. Our results, though modest, may offer a starting point for the use of NO in radiation therapy; however, further research is required to optimize these effects.
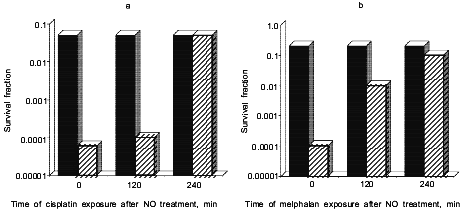
Chemotherapy. Recently, we have examined the effect of NO and reactive nitrogen oxide species (RNOS) on the cytotoxicity of chemotherapeutic agents, with the hope that these results will serve as a guide for delivering NO either in the form of NO donors or through the modulation of NOS. We have found that NO, delivered either by DEA/NO or as a bolus of authentic NO, resulted in increased cytotoxicity of the alkylating agents cisplatin and melphalan (Fig. 3). Further studies showed that V79 cells treated with NO, rinsed, then exposed to the alkylating agent at different times were still sensitive up to several hours after addition of NO. We concluded from this series of experiments that the increase in sensitivity mediated by NO was probably due to inhibition of key DNA repair proteins, perhaps including DNA ligase.
Unlike our studies on the radiosensitization by NO, it appears that enhancement is mediated not by NO directly but through RNOS. This was determined by comparing different NO donor drugs. S-Nitrosothiol complexes were very effective radiosensitizers [47]. However, GSNO and S-nitrosothiols did not enhance the toxicity of either melphalan [46] or cisplatin [45] effectively. A comparison of 2,3-diaminonaphthalene nitrosation following treatment with the various NO donors demonstrated that DEA/NO and PAPA/NO caused significant nitrosative stress, whereas GSNO did not. These results suggest that high cellular levels of NO are required in order to facilitate nitrosation reactions and thereby enhance the cytotoxicity of chemotherapeutic agents, and supports the hypothesis that RNOS are responsible for these effects.Fig. 3. Effect of the NO donor, DEA/NO, on the cytotoxic effect on V79 lung fibroblast cells exposed to cisplatin (a) and melphalan (b) at 0, 120, and 240 min after NO treatment. The black bars correspond to melphalan (4 µg/ml) or cisplatin (25 µg/ml) alone while the stripped bars correspond to 1 mM DEA/NO and melphalan or cisplatin added together. Data from references [45, 46].
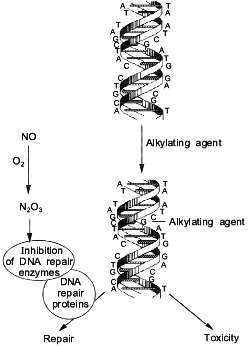
One of the potential targets for RNOS in vivo are the enzymes which collectively make up the cell DNA repair machinery. Some reports have shown that the presence of NO inhibited certain DNA repair proteins. DEA/NO inhibited alkyltransferase [57], formamidopyrimidine-DNA glycosylase (protein fpg) [58], and ligase [59]. However, endonuclease III, endonuclease IV, and uracil glycosylase were not inhibited following exposure to NO at concentrations as high as 10 mM [58]. These findings suggest that the effect of NO is specific to some repair proteins and not others, and that the toxicity of some chemotherapeutic agents which induce certain types of DNA lesions will be enhanced by NO (Fig. 4).
From these studies, it was determined that the intermediates formed from the NO/O2 reaction, and not NO itself, were the chemical species responsible for the inhibition of these DNA repair proteins [57, 58]. Alkyl transferase has critical -SH residues, which upon exposure to aerobic solutions of NO form a S-NO adduct [57]. This adduct inhibited the transfer of an alkyl group from the O6-position of guanine to the thiol residue within the protein, resulting in the potentiation of the toxicity of alkylating agents. We extended these studies to show that H4 hepatoma cells treated with NO donors also exhibited inhibition of alkyl transferase enzyme activity [57]. Furthermore, NO potentiated the cytotoxicity of bis-N,N'-bis(2-chloroethyl)-N-nitrosourea (BCNU), a compound which induces lesions which are repaired by alkyl transferase [57].Fig. 4. Mechanism for enhancement of alkylating agents by NO.
The fpg protein was also inhibited by exposure to NO. This protein is responsible for the repair of lesions such as 8-oxoguanidine and 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine (Fapy) which are caused by oxidative damage [60]. As was the case with alkyl transferase, the intermediates from the NO/O2 reaction were responsible for the inhibition of the enzymatic activity of the fpg protein [58]. This protein is a zinc finger protein with four cysteines [60]; nitrosation of the thiols bound to the zinc metal may form a S-NO adduct and lead to the ejection of the zinc ion. This would result in the loss of protein integrity, thus preventing the protein from binding to DNA. Kroncke et al. also showed that other zinc finger proteins were degraded by the same chemistry as that described above for the fpg protein. Since 1% of the human genome codes for zinc fingers [61], this chemistry provides important insight into the action and biological targets of the NO/O2 reaction.
As we have done for the interaction between NO and radiation, we have begun to examine the effect of NO on the cytotoxicity of melphalan towards two tumors, SCC (solid tumor) and EOC (ascites tumor). Animals bearing SCC tumors and treated with melphalan alone experienced 70% mortality after 50 days, while animals treated with DEA/NO and melphalan together showed 100% mortality over the same time interval. Hence, the presence of DEA/NO appears to inhibit the efficacy of melphalan. We next examined the effect of DEA/NO on the ascites EOC tumor treated with melphalan. Animals treated with melphalan alone experienced 60% mortality after 70 days, while animals treated with melphalan and DEA/NO experienced 35% mortality over the same time interval. These results suggest that DEA/NO improved the effectiveness of melphalan in the ascites tumor.
The major reason for this dichotomy may relate to physiology of NO. Systemic administration of DEA/NO results in hypotension, which reduces blood flow to the solid tumor. This in turn reduces the amount of melphalan delivered to the tumor, thus reducing the efficacy of this drug. The steel effect, which results in the protection of bone marrow against whole body irradiation described above, appears to protect tumors from the toxicity of melphalan. However, ascites tumors may be less subject to the steel effect due to their location in the peritoneal cavity. Thus, enough DEA/NO may reach the peritoneal cavity, thereby possibly mimicking the synergy between DEA/NO and melphalan observed in vitro. These studies suggest that vascular tone is a critical issue when using NO donor compounds in chemotherapy.
Some recent studies have examined the effect of cisplatin [62, 63] and radiation on the expression of iNOS activity [64], and demonstrated that both of these chemotherapeutic agents increase NO production and the expression of iNOS. We (McKinney et al., submitted; Vodovotz et al., manuscript in preparation)and others [65] have demonstrated increased NO production and expression of iNOS following irradiation. Our studies attributed this enhancement in macrophages to increased autocrine production of tumor necrosis factor alpha (TNF-alpha) induced by radiation (McKinney et al., submitted).
Though NO donors and NO inhibitors may not prove as the ultimate single effective therapeutic agent in cancer treatment, they may provide for enhancement of existing and future chemotherapy or radiotherapy regimens. Extensive further work is required, but clinical strategies involving NO may prove useful in cancer treatment.
REFERENCES
1. Hibbs, J. B., Vavrin, Z., and Taintor, R. R.
(1987) J. Immunol., 138, 550-565.
2.Jenkins, D. C., Charles, I. G., Thomsen, L. L.,
Moss, D. W., Holmes, L. S., Baylis, S. A., Rhodes, P., Westmore, K.,
Emson, P. C., and Moncada, S. (1995) Proc. Natl. Acad. Sci.
USA,92, 4392-4396.
3.Thomsen, L. L., Miles, D. W., Happerfield, L.,
Bobrow, L. G., Knowles, R. G., and Moncada, S. (1995) Br. J.
Cancer,72, 41-44.
4. Griffith, O. W., and Stuehr, D. J. (1995) Annu.
Rev. Physiol.,57, 707-736.
5.Nathan, C., and Xie, Q. (1994) J. Biol.
Chem.,269, 13725-13728.
6.Marletta, M. A. (1993) J. Biol.
Chem.,268, 12231-12234.
7.Stuehr, D. J., Abu-Soud, H. M., Rousseau, D. L.,
Feldman, P. L., and Wang, J. (1995) Adv. Pharmacol.,34,
207-213.
8.Marletta, M. A. (1994) Cell,78,
927-930.
9.Wink, D. A., Grisham, M., Mitchell, J. B., and
Ford, P. C. (1996) Meth. Enzymol.,268, 12-31.
10.Wink, D. A., Hanbauer, I., Grisham, M. B., Laval,
F., Nims, R. W., Laval, J., Cook, J. C., Pacelli, R., Liebmann, J.,
Krishna, M. C., Ford, M. C., and Mitchell, J. B. (1996) Curr. Top.
Cell. Regul., 34, 159-187.
11.Stuehr, D. J., and Nathan, C. F. (1989) J.
Exp. Med.,169, 1543-1555.
12.Yim, C. Y., McGregor, J. R., Kwon, O. D.,
Bastian, N. R., Rees, M., Mori, M., Hibbs, J. B. J., and Samlowski, W.
E. (1995) J. Immunol., 155, 4382-4390.
13.Kurose, I., Miura, S., Fukumura, D., Yonei, Y.,
Saito, H., Tada, S., Suematsu, M., and Tsuchiya, M. (1993) Cancer
Res.,53, 2676-2682.
14.Fukumura, D., Yonei, Y., Kurose, I., Saito, H.,
Ohishi, T., Higuchi, H., Miura, S., Kato, S., Kimura, H., Ebinuma, H.,
and Ishi, H. (1996) Hepatology,24, 141-149.
15. Curley, S. A., Roh, M. S., Feig, B., Oyedeji,
C., Kleinerman, E. S., and Klostergaard, J. (1993) J. Leukoc.
Biol.,53, 715-721.
16.Jiang, H., Stewart, C. A., Fast, D. J., and Leu,
R. W. (1992) J. Immunol.,149, 2137-2146.
17.Leu, R. W., Leu, N. R., Shannon, B. J., and Fast,
D. J. (1991) J. Immunol.,147, 1816-1822.
18.Gardner, T. E., Naama, H., and Daly, J. M. (1995)
J. Surg. Res., 59, 305-310.
19.Lejeune, P., Lagade, C. P., Onier, N., Pinard,
D., Ohshima, H., and Jeannin, J. F. (1994) J.
Immunol.,152, 5077-5083.
20.Dinapoli, M. R., Calderon, C. L., and Lopez, D.
M. (1996) J. Exp. Med.,183, 1323-1329.
21.Alleva, D. G., Burger, C. J., and Elgert, K. D.
(1994) J. Immunol., 153, 1674-1686.
22.Vodovotz, Y. (1997) Nitric Oxide: Biol.
Chem.,1, 3-17.
23.Maeda, H., Kuwahara, H., Ichimura, Y., Ohtsuki,
M., Kurakata, S., and Shiraishi, A. (1995) J.
Immunol.,155, 4926-4932.
24.Calderon, C., Huang, Z. H., Gage, D. A.,
Sotomayor, E. M., and Lopez, D. M. (1994) J. Exp.
Med.,180, 945-958.
25.Thomsen, L. L., Lawton, F. G., Knowles, R. G.,
Beesley, J. E., Riveros-Moreno, V., and Moncada, S. (1994) Cancer
Res.,54, 1352-1354.
26.Cobbs, C. S., Brenman, J. E., Aldape, K. D.,
Bredt, D. S., and Israel, M. A. (1995) Cancer Res.,55,
727-730.
27.Prazma, J., Petrusz, P., Mims, W., Ball, S. S.,
and Weissler, M. C. (1995) Otolaryngol. Head Neck
Surg.,113, 541-549.
28.Dong, Z., Staroselsky, A. H., Qi, X., Xie, K.,
and Fidler, I. J. (1994) Cancer Res.,54, 789-793.
29.Xie, K., Huang, S., Dong, Z., Juang, S. H.,
Gutman, M., Xi, E. Q. W., Nathan, C., and Fidler, I. J. (1995) J.
Exp. Med.,181, 1333-1343.
30.Edward, S. P., Cendan, J. C., Topping, D. B.,
Moldawer, L. L., MacKay, S., Copeland, E. M. I. I. I., and Lind, D. S.
(1996) J. Surg. Res.,63, 49-52.
31.Jin, Y., Heck, D. E., DeGeorge, G., Tian, Y., and
Laskin, J. D. (1996) Cancer Res.,56, 1978-1982.
32.Salvemini, D., Seibert, K., Masferrer, J. L.,
Misko, T. P., Currie, M. G., and Needleman, P. (1994) J. Clin.
Invest.,93, 1940-1947.
33.Salvemini, D., Misko, T. P., Masferrer, J. L.,
Seibert, K., Currie, M. G., and Needleman, P. (1993) Proc. Natl.
Acad. Sci. USA,90, 7240-7244.
34.McDaniel, M. L., Kwon, G., Hill, J. R., Marshall,
C. A., and Corbett, J. A. (1996) Proc. Soc. Exp. Biol.
Med.,211, 24-32.
35.Corbett, J. A., Kwon, G., Turk, J., and McDaniel,
M. L. (1993) Biochemistry,32, 13767-13770.
36.Sautebin, L., and Di Rosa, M. (1994) Eur. J.
Pharmacol.,262, 193-196.
37.Laszlo, F., Whittle, B. J., and Moncada, S.
(1994) Br. J. Pharmacol.,113, 1131-1136.
38.Nakatsuka, M., and Osawa, Y. (1994) Biochem.
Biophys. Res. Commun., 200, 1630-1634.
39.Maccarrone, M., Corasaniti, M. T., Guerrieri, P.,
Nistico, G., and Finazzi Agro, A. (1996) Biochem. Biophys. Res.
Commun., 219, 128-133.
40.Maeda, H., Noguchi, Y., Sato, K., and Akaike, T.
(1994) Jpn. J. Cancer Res.,85, 331-334.
41.Nakano, S., Matsukado, K., and Black, K. L.
(1996) Cancer Res.,56, 4027-4031.
42.Orucevic, A., and Lala, P. K. (1996) Cancer
Immunol. Immunother., 42, 38-46.
43.Hubbard, N. E., and Erickson, K. L. (1995)
Cell. Immunol.,160, 115-122.
44.Farias-Eisner, R., Chaudhuri, G., Aeberhard, E.,
and Fukuto, J. M. (1996) J. Biol. Chem.,271,
6144-6151.
45.Wink, D. A., Cook, J. A., Christodoulou, D.,
Krishna, M. C., Pacelli, R., Kim, S., DeGraff, W., Gamson, J.,
Vodovotz, Y., Russo, A., and Mitchell, J. B. (1997) Nitric Oxide
Biol. Chem.,1, 3-17.
46.Cook, J. A., Krishna, M. C., Pacelli, R.,
DeGraff, W., Liebmann, J., Russo, A., Mitchell, J. B., and Wink, D. A.
(1997) Br. J. Cancer,76, 325-334.
47.Mitchell, J. B., Cook, J. A., Krishna, M. C.,
DeGraff, W., Gamson, J., Fisher, J., Christodoulou, D., and Wink, D. A.
(1996) Br. J. Cancer,74, 5181-5184.
48.Howard-Flanders, P. (1957)
Nature,180, 1191-1192.
49.Mitchell, J. B., Wink, D. A., DeGraff, W.,
Gamson, J., Keefer, L. K., and Krishna, M. C. (1993) Cancer
Res.,53, 5845-5848.
50.Bourassa, J., DeGraff, W., Kudo, S., Wink, D. A.,
Mitchell, J. B., and Ford, P. C. I. (1997) J. Am. Chem. Soc.,
119, 2853-2856.
51.Lancaster, J. R., and Hibbs, J. B. (1990)
Proc. Natl. Acad. Sci. USA,87, 1223-1227.
52.Drapiers, J. C., Pellat, C., and Henry, Y. (1991)
J. Biol. Chem., 269, 5127-5131.
53.Vanin, A. F., Mordvintcev, P. I., Hausschildt,
S., and Mulsch, A. (1992) Biochim. Biophys. Acta,1177,
37-42.
54.Liebmann, J., DeLuca, A. M., Coffin, D., Keefer,
L. K., Venzon, D., Wink, D. A., and Mitchell, J. B. (1994) Cancer
Res.,54, 3365-3368.
55.Wood, P. J., Stratford, I. J., Adams, G. E.,
Szabo, C., Thiemermann, C., and Vane, J. R. (1993) Biochem. Biophys.
Res. Commun.,192, 505-510.
56.Ballinger, J. R., Kee, J. W., and Rauth, A. M.
(1996) J. Nucl. Med.,37, 1023-1031.
57.Laval, F., and Wink, D. A. (1994)
Carcinogenesis,15, 443-447.
58.Wink, D. A., and Laval, J. (1994)
Carcinogenesis,15, 2125-2129.
59.Graziewicz, M., Wink, D. A., and Laval, F. (1996)
Carcinogenesis, 17, 2501-2505.
60.O'Connor, T., Graves, R. V., Murcia, G.,
Castaning, B., and Laval, J. (1993) J. Biol. Chem.,268,
9063-9070.
61.Kroncke, K.-D., Fechsel, K., Schmidt, T., Zenke,
F. T., Dasting, I., Wesener, J. R., Bettermann, H., Breunig, K. D., and
Kolb-Bachofen, V. (1994) Biochem. Biophys. Res.
Commun.,200, 1105-1110.
62.Ogura, T., Tatemichi, M., and Esumi, H. (1997)
Biochem. Biophys. Res. Commun.,233, 788-791.
63.Son, K., and Kim, Y. M. (1995) Cancer
Res.,55, 5524-5527.
64.Hatjikondi, O., Ravazoula, P., Kardamakis, D.,
Dimopoulos, J., and Papaioannou, S. (1996) Br. J.
Cancer,74, 1916-1923.
65.Ibuki, Y., and Goto, R. (1997) Free Radic.
Biol. Med.,22, 1029-1035.