Interactions Between Nitric Oxide and Cytochrome P-450 in the Liver
O. Khatsenko
ISIS Pharmaceuticals, Department of Toxicology and Pharmacokinetics, 2292 Faraday Avenue, Carlsbad, CA 92008, USA; fax: (1-760) 603-3862; E-mail: okhatsen@isisph.com
Received February 9, 1998
Bacterial lipopolysaccharide and a diverse array of other immunostimulants and cytokines suppress the metabolism of endogenous and exogenous substances by reducing the activity of hepatic cytochrome P-450 mixed function oxidase system. Although this effect of immunostimulants was first described almost 40 years ago, the mechanism is obscure. Immunostimulants are now known to cause nitric oxide overproduction by cells via induction of nitric oxide synthase. The highly reactive NO radical binds to prosthetic groups such as heme or iron-sulfur clusters leading to either activation or (more often) inhibition of iron-containing enzymes. It has been known for years that NO also binds to the heme moiety of cytochrome P-450 (CYP) with high affinity. However it was only recently demonstrated that binding of NO to CYPs also inhibits their enzymatic activity. This applies to both exogenously derived as well as endogenously synthesized NO. Suppression of CYP-dependent metabolism, which is a major problem of inflammatory liver diseases, can be significantly reversed by inhibition of NO synthesis in vivo under experimental conditions. The present paper reviews the findings implicating NO as a major factor mediating the suppression of CYP expression caused by endotoxins and immunostimulants in general. NO-mediated suppression of the metabolism of endogenous and exogenous substances under inflammatory conditions may contribute to the clinical manifestations and may be an important consideration for rational drug therapy in these conditions.
KEY WORDS: nitric oxide, cytochrome P-450, liver, rat, mice, inflammation, cytokines, bacterial lipopolysaccharide, endotoxin, Chlamydia trachomatis infection
To date a substantial amount of evidence that infections can alter the biotransformation of drugs and chemicals in the liver has accumulated. Many agents, both biological and chemical, which have the ability to alter the immune system or stimulate host defense mechanisms, also have the ability to impair processes involved in drug metabolism [1, 2]. Today, the list of immunomodulators which have been reported to alter cytochrome P-450 mediated metabolism in animals includes vaccines, attenuated bacteria, nonspecific immunostimulants, viral compounds affecting the reticuloendothelial system (RES), bacterial lipopolysaccharide (LPS), interferon (IFN) inducers, and some cytokines. All of these agents are effective in depressing the microsomal mixed function oxidase system when administered in vivo but have no effect when added in vitro to the isolated preparations of hepatic microsomes. Since the first reports, numerous studies revealed that such suppression in drug metabolism occurs in more than one animal species (including rats, mice, rabbits, and humans) and affects more than one of the cytochrome P-450 molecular families and their individual members.
Soon after the first report of impaired theophylline elimination during influenza in humans [3], it was proposed [4] that during infectious disease the formation of interferon (IFN) causes a decrease in the amount of cytochrome P-450 in the liver available for the metabolism of the drug in patients. IFN has been proposed to be the principal mediator of the suppression of cytochrome P-450 microsomal monooxygenase by immunostimulants. However, some caution was warranted later in implicating IFN per se as the initiating agent responsible for the depression of oxidative drug metabolism that follows immune system perturbation [5, 6].
Lately, attention has focussed upon the possible role of cells of the RES and, in particular, their secretory products as mediators of the loss of cytochrome P-450. Much of this work stems from the many earlier reports that LPS, a well-known activator of macrophages, invariably produced a marked depression of hepatic cytochrome P-450 levels. Although it was apparent from such reports that LPS administration could perturb normal drug metabolizing capacity, the mechanism by which the effect was elicited was not clear. Another series of experiments [7] demonstrated that cells of the RES, such as peritoneal macrophages or Kupffer cells, following their activation with recognized RES-active agents, could release a factor that was able to interact directly with hepatocytes in culture to depress intra-cellular levels of active cytochrome P-450. However, at this stage the factor was not identified.
It was not long before interleukin (IL)-1 was considered a possible candidate for the macrophage or Kupffer cell factor affecting the levels of cytochrome P-450 in isolated hepatocyte culture [8]. In 1986 Ghezzi et al. [9] also reported that another macrophage-derived cytokine, recombinant tumor necrosis factor (TNF), could alter cytochrome P-450 levels and associated drug metabolism. However, the same group later discovered that while LPS, IL-1, or TNF all caused depression of cytochrome P-450 in mice, only IL-1 depressed cytochrome P-450 in cultured hepatocytes. The authors suggested that LPS- and TNF-stimulated monocytes released a factor capable of depressing cytochrome P-450 in cultured hepatocytes. Recently, several groups of researchers have published results on the inhibition of cytochrome P-450-dependent metabolism by another member of the interleukin family, IL-6 [10]. However, again, to explain the obvious contradiction of the IL-6 data from in vitro and in vivo experiments, the authors suggested the involvement of a second, distinct mechanism.
Nitric oxide (nitrogen monoxide, .N=O), a simple and relatively unstable under aerobic conditions radical has been identified in recent years as a potent and pleiotropic mediator (for review see [11, 12]). NO is synthesized from L-arginine in process catalyzed by NO synthase (NOS) and inhibited by certain analogs of L-arginine, including NG-monomethyl-L-arginine (L-NMMA), NG-nitro-L-arginine methyl ester (L-NAME), N-iminoethyl-L-ornithine (L-NIO), etc. An inducible NOS (iNOS) can be expressed in a wide variety of cells, including vascular smooth muscle cells, cardiac myocytes, and immune cells such as macrophages and neutrophils [13]. Liver cells like hepatocytes and Kupffer cell were also shown to synthesize NO [12, 13].
Nitric oxide has a great affinity for iron, either of heme or non-heme origin. The affinity constant of NO for hemoglobin (Hb), measured at half-saturation, is 3·1010 M-1 which is 1000-fold greater than that of CO, while the affinity constant of O2 is only 6·104 M-1 [14]. HbNO reacts with O2 to give rise to Hb(III). NO can also bind loosely to heme-iron(III) and reduce it slowly to heme-iron(II).
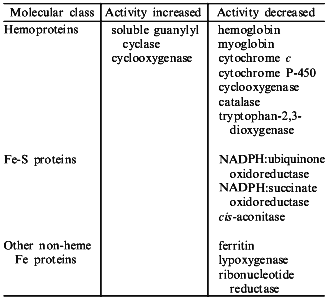
NO binds to many different types of hemoproteins (the table). NO is a high affinity inhibitor of many oxidases and oxygenases through its binding, in place of oxygen, to hemes of type a (cytochrome oxidase of E. coli), b (catalase, peroxidases, tryptophan-2,3-dioxygenase, cytochrome P-450, etc.), or d1 (nitrate reductase) giving characteristic EPR spectra of nitrosyl-hemoproteins; it also binds to cytochrome c. NO also binds to non-heme iron containing proteins (transferrin, ferritin, lipoxygenase, rhodopsin) as well as to iron-sulfur and multi-copper proteins [14]. Binding of NO to heme and non-heme iron leads to either inhibition or (more rarely) activation of iron-containing enzymes (the table). The inhibitory effect of NO seems to derive from: 1) the capability of NO to reduce the ferric enzyme to the ferrous form, which is inactive; 2) competition for the iron sites available for endogenous (e.g., O2) as well as exogenous ligands; and 3) the radical scavenging ability of the nitroxide radical.
Iron-containing proteins which are known to be molecular targets for NO
INHIBITION OF CYTOCHROME P-450 BY NO IN VITRO
The use of nitric oxide as a spin label to probe the electronic structure of the heme prosthetic groups in both native and structurally perturbed hemoproteins has received much attention for the last 30 years [15, 16]. Hemoglobin, myoglobin, as well as horseradish peroxidase and cytochrome c oxidase form stable ferrous NO complexes [17]. The first reports suggesting that microsomal and submicrosomal cytochrome P-450 forms both ferric- and ferrous-NO complexes were as early as 1968 [15, 18]. In 1975 Ebel et al. [19] and in 1978 O'Keeffe et al. [20] in detailed studies demonstrated, using rat microsomal P-450 and P-450cam from P. putida, that the ferrous P-450--NO complex is unstable and is converted to a specie which has an EPR spectrum similar to that of ferrous P-420--NO (with a symmetrical signal at g = 2.04). The spectrum of the ferrous-NO complex of cytochrome P-450cam resembles that of its ferrous-CO complex [19]. It appears that the optical absorbance spectral properties of both the ferric and ferrous-NO complexes of cytochromes P-450 are unique relative to those of other hemoproteins, e.g., red-shifted Soret band maxima for both the ferric- and ferrous-NO complexes. This suggests that the interaction which gives rise to the 450 nm Soret absorbance band maximum of the ferrous-CO complex is present in both oxidation states of the enzyme at least when nitric oxide is bound. Thus, not only CO and O2 [21] but also NO are directly competitive with each other for binding to the heme iron [20].
The first evidence that binding of NO to microsomal P-450s in vitro leads, in fact, to inhibition of their enzymatic activities was provided simultaneously by Wink et al. [22] and Khatsenko et al. [23, 24]. In the experiments of Wink et al., a saturated NO solution and NO derived from the chemical decomposition of an NO adduct of diethylamine, Na[Et2NN(O)NO] (DEA/NO), inhibited CYP1A-dependent O-deethylation of 7-ethoxyresorufin (EROD) and CYP2B-dependent O-dearylation of 7-benzyloxyresorufin (BROD) activities of rat hepatic S-9 or microsomal fractions or purified rat liver CYP2B1. The authors observed two distinct inhibitory phases in all experiments (reversible and irreversible), regardless of whether NO was added prior to initiation of the reactions with NADPH or during the course of substrate turnover.
To demonstrate that the irreversible phase of inhibition is caused by NO, these workers were able to abrogate this inhibition by adding BSA, a known scavenger of NO. Similarly, in our earlier studies treatment of hepatic microsomes with NO produced by chemical decomposition of 3-morpholinosydnonimine (SIN-1) or by nitric oxide synthase substantially suppressed P-450-dependent oxygenation reactions [24]. This effect for NO was seen with hepatic microsomes prepared from two species (rat and chicken) and belonged to two distinct families of P-450 molecular isoforms (CYP1A and CYP2B).
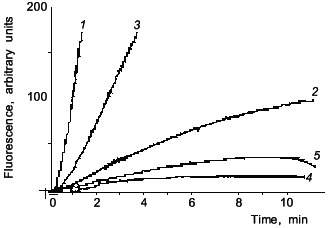
Figure 1 shows data from a representative experiment on NO inhibition of CYP2B-mediated 7-pentoxyresorufin O-dealkylation (PROD) activity in phenobarbital (PB)-induced rat liver microsomes. Likewise, with BSA, addition of Mb (curve 3) preserved 7-PROD activity from inhibition caused by SIN-1. Since SIN-1 is also known to produce O2- radical, SOD was used to assure that observed inhibition is indeed due to NO. Our spectral studies confirmed that NO reacts in vitro with both Fe2+- and Fe3+-hemes in microsomal P-450s [24]. Also, in accord with Wink et al., it was found that CYP1A activity is ~7 times less sensitive than 7-PROD towards inhibition by NO (the IC50 determined by Wink et al. was 8.2 µM for BROD, whereas for EROD it was 58 µM NO). These results suggested that though NO may be a nonspecific inhibitor of all cytochromes P-450, it could affect cytochrome P-450 forms differentially, based, perhaps, on differences in accessibility of heme to NO.
Since the first reports there has been a growing number of publications indicating the role of NO in the suppression of cytochrome P-450-dependent metabolism under a variety of different in vitro as well as in vivo conditions. Stadler et al. [25] reported a study on the inhibition of members of the CYP1A subfamily by nitric oxide. In one of their experimental approaches, these workers used V79 Chinese hamster cells genetically engineered for stable expression of rat and human CYP1A1 and CYP1A2. They found that incubation of these cells with sodium nitroprusside (SNP) or S-nitrosoacetyl-penicillamine (SNAP) led to a concentration-dependent inhibition of all four tested enzymes. CYP1A1-mediated aryl hydrocarbon hydroxylase (AHH) was more susceptible to the inhibitory effect of NO than CYP1A2-mediated EROD, and this difference was more pronounced with rat than with human enzymes. Thus, these authors have demonstrated differences in the susceptibility to NO, not only among different species (rat and human), but also among the forms of one P-450 subfamily (CYP1A1 and CYP1A2).Fig. 1. Fluorescence curves of resorufin formation from 7-pentoxyresorufin: effects of SIN-1, myoglobin, and SOD. Microsomes from rats treated with PB (80 mg/kg for 54 h) were assayed for PROD activity (1); 2) 5 mM SIN-1 was added prior to start of the reaction with NADPH; 3) 20 µM myoglobin was present in the incubation mixture prior to addition of 5 mM SIN-1; 4) 10 mM SIN-1 was added prior to start of reaction with NADPH; 5) 200 units SOD were present prior to the addition of 10 mM SIN-1.
Stadler at al. have also demonstrated that endogenous NO produced by hepatocytes stimulated with a combination of rmTNF-alpha, rhIL-1, rrIFN-gamma, and LPS inhibited completely the AHH activity of control and beta-NF-induced cells [24]. L-NMMA significantly attenuated the reduction in CYP1A1-mediated benzo[a]pyrene turnover in cells stimulated for NO production. Osawa et al. [26] have accessed the activities of two constitutive P-450 isoforms expressed in primary hepatocytes and hydroxylating testosterone in different positions of the steroid ring: 2alpha- and 16alpha- (CYP2C11) and 6beta- (CYP3A2). Treatment of cells with LPS and cytokines lead to 43, 55, and 33% decreases in 2alpha-, 16alpha-, and 6beta-hydroxylase activities, respectively. The partial reversal of this inhibition by L-NMMA led the authors to suggest that other NO-independent mechanisms may also be involved. Long-term effects of NO in the design of the experiments prevailed over short-term effects enabling Osawa et al. to speculate about the primary role of non-reversible inhibition. However, based on conclusions of Stadler et al., these researchers did not recognize the possible role of NO in the down-regulation of P-450 gene expression. On the other hand, it is well documented that suppression of P-450-mediated activities is well correlated with the decrease in corresponding mRNA and protein levels [1, 2]. Recently, Carlson and Billing [27] published results of the detailed study on the role of NO in cytokine-mediated regulation of cytochrome P-450 in vitro. The authors accessed P-450 content and several CYP protein levels in primary hepatocytes treated with cytokine combination consisting of TNF-alpha, IL-1beta, and IFN-gamma. The combination was found to suppress P-450 content by 69%. Treatment with cytokines also resulted in a decrease of each CYP isozyme, with CYP2B exhibiting the greatest loss (to 33% of untreated cell levels) as did the treatment with NO donor. TNF-alpha and IL-1beta caused the induction of NO followed by the decrease in CYP2B1/2 and CYP3A2 proteins (TNF-alpha) and CYP1A2, CYP2B1/2, CYP2C11, and CYP3A2 proteins (IL-1beta), while IL-6 suppressed these P-450s without inducing NO synthesis. In these experiments addition of L-NMMA and L-NAME resulted in almost complete protection of the activity of every P-450 isoform studied (up to 78-99% of control). Moreover, L-NMMA completely protected all P-450 apoproteins from the suppression by IL-1beta (95-100%) and almost completely protected from TNF-alpha (89-96%). It was concluded, therefore, that down-regulation of P-450 by TNF-alpha and IL-1beta in vitro is directly associated with NOS induction, whereas in the case of IL-6 these pathways are separated. In addition, NO was shown to mediate the suppression of P-450 metabolism in hepatocytes by IFN [28]. Another piece of evidence in favor of the NO role came from experiments on the suppression of P-450-dependent vasodilatation by LPS in isolated rat kidney, thus demonstrating that this effect of NO is not limited to liver cells [29].
ROLE OF NO IN INHIBITION OF CYTOCHROME P-450 IN VIVO
Physiological concentrations of endogenously generated NO as high as 0.075 µM have been reported for stimulated cerebellar slices [30]. Murine peritoneal macrophages in culture, activated with LPS, synthesized approximately 150 nmoles nitrate/106 cells per 24 h [31]. Curran and coworkers [32] have demonstrated that NO is generated from L-arginine by hepatic parenchymal cells (hepatocytes) in quantities even higher than those generated by activated immune cells, endothelial cells, and neutrophils (2·105 cells/ml in the presence of supernatant from LPS-activated Kupffer cells produce up to 0.74 mmole nitrate/nitrite per 18 h). These findings, together with those of Wink et al. [22] of complete reversible P-450 inhibition at steady-state NO concentrations >0.07 µM, imply that the quantities of NO produced in vivo are sufficient to affect cytochrome P-450 and indicate a possible role for NO in the regulation of P-450 activity in vivo.
In vivo induction of hepatic NO synthesis was reported in rodents following endotoxemia and gram negative sepsis as well as injection of inactivated Corynebacterium parvum and several other infectious agents. In animals treated with C. parvum or/and LPS, inhibition of NO biosynthesis results in liver damage [33]. On the other hand, the induction of NO synthesis in hepatocytes was shown to accompany a profound inhibition of total cellular protein synthesis [34]. However, the inhibition of cytochrome P-450 is unlikely to be related to nonspecific protein inhibition in the liver by NO. Using electron paramagnetic resonance (EPR), Chamulitrat et al. [35] were able to identify cytochrome P-450 and P-420 as intracellular targets of NO in the livers of mice. They consistently found a suppression of EPR signals attributable to ferric low-spin cytochrome P-450/P-420 peaks in the livers of mice treated with C. parvum and C. parvum + LPS. We also demonstrated earlier that NO induced by LPS in rats is able to depress P-450 heme as well as total microsomal heme and cytochrome b5 contents [24, 36]. This notion was also confirmed by Kim et al. [37] using microsomes from rats treated with LPS and LPS + L-NMMA and LPS + aminoguanidine (AG). Similar data were also obtained by the same workers using freshly isolated hepatocytes from C. parvum-treated rats as wells as with cultured hepatocytes treated with cytokines [37, 38]. We hypothesized then that NO could inhibit spectral detection of total P-450 heme by preventing formation of carbon monoxy complex [24]. However, the loss of total extractable microsomal heme that parallels the decrease in P-450 heme as well as possibility of reconstitution of P-450 activity let Kim et al. argue in favor of true loss of P-450 heme [38].
In addition, we were able to demonstrate later that NO can suppress cytochrome P-450 gene expression in vivo, which is believed to be the major mechanism of down-regulation of this enzyme under inflammatory conditions. Likewise, in in vitro studies of Stadler et al. [25] and Carlson and Billing [27], we found that NO is mainly responsible for the suppression of synthesis of at least several constitutive rat P-450 isozymes in vivo [36]. For instance, treatment of rats with LPS caused substantial (40-45%) loss in CYP2C11 and CYP3A2 mRNAs followed by more profound decrease in corresponding apoproteins (52 and 81%, respectively). However, cotreatment of animals with L-NAME resulted in from 29% (3A2) to 100% (2C11) protection of the corresponding apoproteins from suppression by LPS. A similar picture was observed with constitutively present CYP2B isoforms. Moreover, systemic infection with intracellular parasite Chlamydia trachomatis resulted in suppression of P-450-mediated drug metabolism in mice [39]. Six days after inoculation with C. trachomatis, CYP1A and CYP2B-mediated metabolism in the livers of the mice was diminished to 49% of control levels, whereas in L-NAME-treated animals the decrease in the activities was largely blocked. These results suggest that infections can depress cytochrome P-450 in a manner similar to other inflammatory conditions and that NO is also a mediator of this depression.
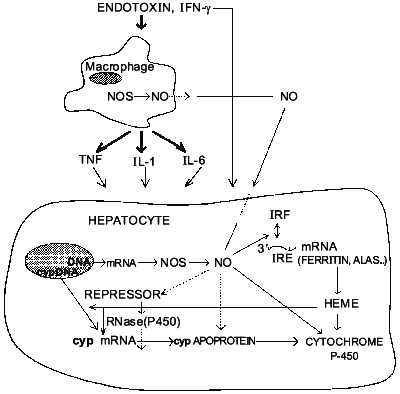
NO also inhibits inducible P-450 isoforms in vivo. For instance, administration of LPS decreased total P-450 content and metabolism of 7-pentoxyresorufin [23] as well as ethylmorphine and midazolam [40] in PB-treated rats. Inhibition of NO formation by L-NAME, L-NMMA, or AG prevents these effects of endotoxin. Furthermore, upon induction with PB, CYP2B1/2 mRNA and protein express equally large susceptibility towards inhibition by LPS (up to 75%) [41]. And, like with constitutive isoforms, PB-inducible ones were largely protected by treatment with the NOS inhibitor L-NAME. Therefore, in contrast to conclusions of Stadler et al. [25] it seems conceivable that NO can indeed regulate the expression of particular cytochrome P-450 isoforms at least at the pretranslational level. This, to our knowledge, was the first example of possible regulation of specific protein expression by NO in vivo. Interestingly, using two distinct schemes of CYP2B induction by PB and NO induction by LPS, we hypothesized that NO likely affects inducible P-450s post-transcriptionally [41], perhaps by altering the stability of its mRNA. The mechanism by which NO can affect mRNA stability is not clear. However, it is known that NO can activate the binding of iron-responsive factor (IRF) to iron-responsive element (IRE) present in the 5'-untranslated region of mRNA for ferritin (a major intracellular iron storage protein) and for delta-aminolevulinate synthetase (ALAS, which catalyzes the rate-determining step in heme biosynthesis) and in the 3'-untranslated region of the mRNA for transferrin receptor (which is the major mechanism for the uptake of iron) [42, 43]. It, therefore, seems feasible that intracellular heme may act as a second mediator of NO-dependent suppression of P-450 gene expression. However, despite the evidence that heme can down-regulate P-450 levels [44], other reports contradict this view [45, 46]. Thus, presently, the role of heme in regulation of cytochrome P-450 gene expression remains controversial. Nevertheless, it is possible to speculate now that NO may inhibit cytochrome P-450 via interplay of two mechanisms. It has an immediate effect on P-450 catalytic activity by binding to P-450 heme and secondary long-term effect on suppression of P-450 via gene inhibition. Figure 2 summarizes the possible interactions between L-arginine-NO and cytochrome P-450 pathways in the liver.
However, the failure of several other groups of investigators to implicate NO as a mediator of the inhibition of cytochrome P-450 in vitro [47, 48] and in vivo [49] requires further studies directed toward depicting the differences in the models used by those investigators. It is likely, however, that NO is not the only mediator of the down-regulation of hepatic drug-metabolizing system during inflammation (other mediators, like IL-6 may act NO-independently), but it is an important molecule whose overproduction during endotoxin shock or infection could have significant consequences on regulation of drug metabolism under these conditions.Fig. 2. Postulated sites of interactions between L-arginine-NO and cytochrome P-450 pathways in rodent liver.
REFERENCES
1.Renton, K. W. (1986) Clin. Biochem.,
19, 72-75.
2.Moochhala, S. M. (1991) Ann. Acad. Med.
Singapore, 20, 13-18.
3.Chang, K. C., Bell, T. D., Lauer, B. A., and Chai,
H. (1978) Lancet, 1, 1132-1133.
4.Renton, K. W., Deloria, L. B., and Mannering, G. J.
(1978) Mol. Pharmacol., 14, 672-681.
5.Mannering, G. J., Renton, K. W., El-Azhary, E., and
Deloria, L. B. (1980) Ann. N. Y. Acad. Sci., 350,
314-331.
6.Parkinson, A., Lasker, J., Kramer, M. J., Huang, M.
T., Thomas, P. E., Ryan, D. E., Reik, L. M., Norman, R. L., Levin, W.,
and Conney, A. H. (1982) Drug Metab. Dispos., 10,
579-585.
7.Peterson, T. C., and Renton, K. W. (1984) J.
Pharmacol. Exp. Ther., 229, 299-304.
8.Ghezzi, P., Saccardo, B., Villa, P., Rossi, V.,
Bianchi, M., and Dinarello, C. A. (1986) Infect. Immun.,
54, 837-840.
9.Ghezzi, P., Saccardo, B., and Bianchi, M. (1986)
Biochem. Biophys. Res. Commun., 136, 316-321.
10.Williams, J. F., Bement, W. J., Sinclair, J. F.,
and Sinclair, P. R. (1991) Biochem. Biophys. Res. Commun.,
178, 1049-1055.
11.Vane, J. R. (1994) Phil. Trans. Roy. Soc.
Lond. B., 343, 225-246.
12.Wolin, M. S., and Gross, S. S. (1995) Annu.
Rev. Physiol., 57, 737-769.
13.Moncada, S., Palmer, R. M. G., and Higgs, E. A.
(1991) Pharmacol. Rev., 43, 109-142.
14.Henry, Y., Ducrocq, C., Drapier, J. C., Servent,
D., Kruszyna, R., Smith, R., and Wilcox, D. (1991) Eur. Biophys.
J., 20, 1-15.
15.Miyake, Y., Gaylor, J. L., and Mason, H. S.
(1968) J. Biol. Chem., 243, 5788-5797.
16.Saprin, A. N., Ramseyer, J., McConn, J., and
Piette, L. H. (1977) Biochem. Biophys. Res. Commun., 77,
789-796.
17.Kon, H. (1968) J. Biol. Chem., 243,
4350-4357.
18.Ullrich, V., Cohen, B., Cooper, D. Y., and
Estabrook, R. W. (1968) in Structure and Functions of Cytochromes
(Okunuki, K., Kamen, M. D., and Sekuzu, I., eds.) University Press,
Baltimore, pp. 649-655.
19.Ebel, R. E., O'Keeffe, D. H., and Peterson, J. A.
(1975) FEBS Lett., 55, 198-201.
20.O'Keeffe, D. H., Ebel, R. E., and Peterson, J. A.
(1978) J. Biol. Chem., 253, 3509-3516.
21.Peterson, J. A., Ishimura, Y., and Griffin, B. W.
(1972) Arch. Biochem. Biophys., 149, 197-208.
22.Wink, D. A., Osawa, Y., Darbyshire, J. F., Jones,
C. R., Eshenauer, S. C., and Nims, R. W. (1993) Arch. Biochem.
Biophys., 300, 115-123.
23.Khatsenko, O. G., Gross, S. S., Mitchell, J. A.,
Rifkind, A. R., and Vane, J. R. (1992) Br. J. Pharmacol.,
107, P196.
24.Khatsenko, O. G., Gross, S. S., Rifkind, A. B.,
and Vane, J. R. (1993) Proc. Natl. Acad. Sci. USA, 90,
11147-11151.
25.Stadler, J., Trockfeld, J., Schamalix, W. A.,
Brill, T., Siewert, J. R., Greim, H., and Doehmer, J. (1994) Proc.
Natl. Acad. Sci. USA, 91, 3559-3563.
26.Osawa, Y., Davila, J. C., Nakatsuka, M., Meyer,
C. A., and Darbyshire, J. F. (1995) Drug Metab. Rev., 27,
61-72.
27.Carlson, T. J., and Billings, R. E. (1996)
Mol. Pharmacol., 49, 796-801.
28.Donato, M. T., Guillin, M. I., Jover, R.,
Casteli, J. V., and Gumer-Lechan, M. I. (1997) J. Pharm. Exp.
Ther., 291, 484-490.
29.Oyekan, A. O. (1995) Eur. J. Pharmacol.,
277, 123-132.
30.Shibuki, K., and Okada, D. (1991) Nature,
349, 326-328.
31.Stuehr, D. J., and Marletta, M. A. (1987)
Cancer Res., 47, 5590-5594.
32.Curran, R. D., Billiar, T. R., Stuehr, D. J.,
Hofmann, K., and Simmons, R. L. (1989) J. Exp. Med., 170,
1769-1774.
33.Billiar, T. R., Curran, R. D., Harbrecht, B. G.,
Stuehr, D. J., Demetris, A. J., and Simmons, R. L. (1990) J.
Leukocyte Biol., 48, 565-569.
34.Billiar, T. R., Curran, R. D., West, M. A.,
Hofmann, K., and Simmons, R. L. (1989) Arch. Surg., 124,
1416-1421.
35.Chamlitrat, W., Jordan, S. J., Mason, R. P.,
Litton, A. L., Wilson, J. G., Wood, E. R., Wolberg, G., and Molina de
Vedia, L. M. (1995) Arch. Biochem. Biophys., 316,
30-37.
36.Khatsenko, O. G., and Kikkawa, Y. (1997) J.
Pharm. Exp. Ther., 280, 1463-1470.
37.Kim, Y-M., Bergonia, H. A., Muller, C., Pitt, B.
R., Watkins, W. D., and Lancaster, J. R. (1995) Adv. Pharmacol.,
34, 277-291.
38.Kim, Y-M., Bergonia, H. A., Muller, C., Pitt, B.
R., Watkins, W. D., and Lancaster, J. R. (1995) J. Biol. Chem.,
270, 5710-5711.
39.Khatsenko, O. G., Barteneva, N. S., de la Maza,
L., and Kikkawa, Y. (1998) Biochem. Pharmacol., in press.
40.Muller, C. M., Scierka, A., Stiller, R. L., Kim,
Y-M., Cook, D. R., Lancaster, J. R., Buffington, C. W., and Watkins, W.
D. (1996) Anesthesiology, 84, 1435-1442.
41.Khatsenko, O. G., Gross, S. S., and Boobis, A. B.
(1997) Tox. Lett., 90, 207-216.
42.Drapier, J. C., Hirling, H., Wietzerbin, J.,
Kaldy, P., and Kuhn, L. C. (1993) EMBO J., 12,
3643-3649.
43.Weiss, G., Goossen, B., Doppler, W., Fuchs, D.,
Pantopoulos, K., Werner-Felmayer, G., Wachter, H., and Hentze, M. W.
(1993) EMBO J., 12, 3651-3657.
44.Srivastava, G., Hansen, A. J., Bawden, M. J., and
May, B. K. (1990) Mol. Pharmacol., 38, 486-493.
45.Padmanaban, G., Venkateswar, V., and Rangarajan,
P. N. (1989) Trends Biochem. Sci., 14, 492-496.
46.Sinclair, P. R., Bement, W. J., Haugen, S. A.,
Sinclair, J. F., and Guzelian, P. S. (1990) Cancer Res.,
50, 5219-5224.
47.Monshouwer, M., Witkamp, R. F., Nijmeijer, S. M.,
van Amsterdam, J. G., and van Miert, A. S. (1996) Tox. Appl.
Pharm., 137, 237-244.
48.Sewer, M. B., and Morgan, E. T. (1997)
Biochem. Pharmacol., 54, 729-737.
49.Hodgson, P. D., and Renton, K. W. (1995) Int.
J. Immunopharmacol., 17, 995-1000.