REVIEW: Stress, Adaptation, and Nitric Oxide
I. Yu. Malyshev* and E. B. Manukhina
Institute of General Pathology and Pathophysiology, Russian Academy of Medical Sciences, Baltiiskaya ul. 8, Moscow, 125315 Russia; fax: (095) 151-0421; E-mail: nii@pathophys.msk.ru* To whom correspondence should be addressed.
Received October 27, 1997
The biological role of nitric oxide (NO) has been studied for more than ten years. Nevertheless, the number of investigations in this field continues to increase. It is now suggested that NO is a previously unrecognized, very important regulator of physiological functions and cell metabolism in the body. Through the application of the methods of molecular biology, more and more data are being accumulated on the regulatory role of NO in the mechanism of gene expression and protein biosynthesis. The data presented in this review show an important role of NO in stress and adaptive responses of organisms and thereby expand existing notions on the biological role of this unique molecule. This review substantiates the idea that the system of NO generation is a newly discovered stress-limiting system. The action of this NO-ergic system is based on the capability of NO to limit key links of the stress reaction and to enhance the potency of endogenous defense systems of the organism. The role of NO is considered at the major stages of adaptation: 1) at the urgent stage related with the stress reaction; 2) at the stage of the transition from urgent to long-term adaptation; and 3) at the stage of long-term adaptation characterized by the formation of stable protective effects. It is demonstrated that pharmacological "imitation" of the activated NO-ergic system by administration of NO donors to the organism provides in many instances an efficient protection against stress damage and enhances the adaptive capacity of the organism.
KEY WORDS: stress, stress-limiting system, stress system, urgent and long-term adaptation, adaptive defense, nitric oxide, NO-synthase, NO-synthase inhibitors, NO donors, stress proteins, gene expression
All living organisms, from prokaryotes to higher eukaryotes, respond to stress changes of the environment in strikingly various ways. However, with all the diversity of stress responses both at the cell level and at the level of the organism, the major evolutionary task they are intended to solve is increasing the organism's resistance to the influencing factor, that is, the development of adaptation.
According to a concept developed by Meerson [1], the formation of protective effects of adaptation is provided by activation of the genetic apparatus and changes in cell metabolism along with changes in the functioning of virtually all major systems of the organism: nervous, endocrine, cardiovascular, respiratory, muscular, etc. It is obvious, therefore, that the most important role in the development of adaptation belongs to universal factors controlling the physiological systems and gene expression.
According to current ideas, one such universal regulator is nitric oxide (NO). NO is synthesized in the organism from L-arginine by the enzyme NO-synthase. NO participates in a great number of physiological processes: signal transmission in the brain; regulation of cardiovascular, immune, gastrointestinal, and urogenital systems [2-4]. From the early 1990s very important proofs have appeared indicating that NO is involved in the regulation of the genetic apparatus both at the level of transcription factors [5] and at the level of transcription mechanisms as such [6, 7] and mRNA translation [8].
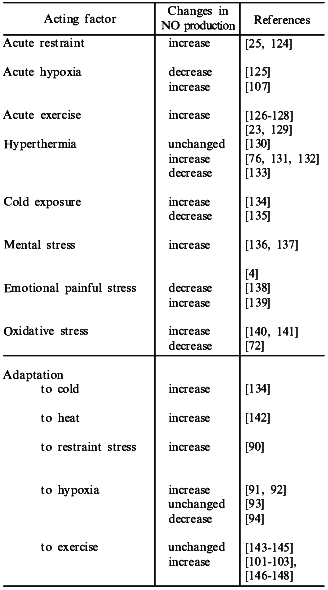
Finally, the data that NO production may be considerably modified in stress and during adaptation to diverse factors (the table) have immediately entailed a hypothesis that NO plays an important role in stress and adaptive responses of the organism. The present review is devoted to analysis of this problem.
Nitric oxide production in various types of stress and adaptation
1. THE ROLE OF NO IN STRESS REACTIONS
The scheme of Fig. 1 summarizes the available data on the development of a stress reaction and shows possible NO-dependent mechanisms of this process. It is seen that any strong impact of the environment such as emotional, painful, or immobilization stresses, acute hypoxia, severe exercise, or high temperature or cold induces a standard stress reaction. The stress reaction is characterized by definite behavioral responses, such as alertness, suppression of sexual and feeding behavior, and the increased production and release of stress hormones, i.e., glucocorticoids, catecholamines, prolactin, growth hormone, etc. [9].
Intensity of the stress reaction is determined by the relationship between the activation of the stress system by the organism's response to the stressor and the activation of "stress-limiting systems", which can restrict excessive activation of the stress system and, thereby, the detrimental effect of stress hormones [9-12].Fig. 1. Stress reaction and the participation of NO in its regulation (see text for explanations). Double light arrow, activating effect of NO; dark arrow, inhibiting effect of NO.
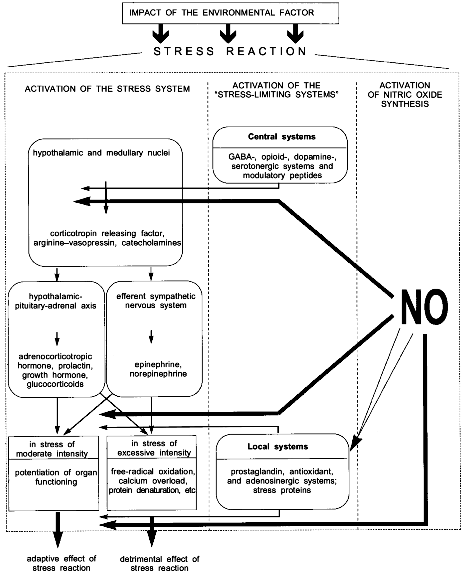
The stress system has two major divisions: central and peripheral [9]. The central division is represented by the medullary and hypothalamic nuclei whose neurons release corticotropin releasing factor (CRF), catecholamines, and arginine--vasopressin. The peripheral division is represented by the hypothalamic--pituitary--adrenal axis and the efferent sympathetic nervous system [13]. Key metabolites of the stress system peripheral division are adrenocorticotropic hormone (ACTH), prolactin, growth hormone, glucocorticoids, norepinephrine, and epinephrine. Regulation of the stress system is extremely complicated and occurs at multiple levels. It is provided by direct neurohumoral influences and negative feedback autoregulation at both the central and peripheral levels [9]. For instance, CRF inhibits CRF-neurons whereas norepinephrine via collateral fibers and prejunctional receptors inhibits catecholaminergic neurons [14]. In addition, CRF-neurons and adrenergic neurons are stimulated by serotonin and acetylcholine and inhibited by glucocorticoids, gamma-aminobutyric acid (GABA), ACTH, and opioid peptides [15-17].
In a short-term exposure to stress of moderate intensity inducing, correspondingly, an appropriate stress reaction, the release of stress hormones results in potentiation of organ functioning and overall mobilization of the organism. However, if the stress reaction is excessively intense or prolonged and is accompanied by a considerable release of stress hormones, cells become exposed to activated free-radical oxidation, intracellular calcium overload, suppressed energy production, decreased synthesis of most proteins, and protein structure denaturation. These exert a detrimental action on organs and tissues and, thereby, the stress reaction is transformed from a link of adaptation into a link of pathogenesis of various diseases [18].
Stress-limiting systems counteract the excessive activation of the stress system. Meerson [10, 12, 19] has described five following principal features characteristic of stress-limiting systems: 1) ability to be activated by stress, i.e., stress-inducibility; 2) ability to restrict release and/or production of stress hormones; 3) ability to restrict stress damage; 4) ability of exogenous metabolites of stress-limiting systems to increase and ability of inhibitors of these systems to decrease both the organism's resistance to stress and the adaptive capacity of the organism; and 5) ability to enhance its own activity and/or reactivity in the process of adaptation to repeated action of environmental factors.
In full consistency with the stress system arrangement (central and peripheral divisions), stress-limiting systems are divided into central ones which primarily restrict the activity of the stress system central link, and local ones which enhance the resistance of cell structures and organs to injury [10, 19] (Fig. 1). The central stress-limiting systems include the GABA-ergic, opioidergic, dopaminergic, and serotonergic systems and some modulatory peptides such as substance P, enkephalin, beta-endorphin, etc. Local protective systems include the prostaglandin, antioxidant, adenosinergic systems and the system of stress proteins Hsp70 [19-21].
The activation of stress-limiting systems coupled with the activation of stress-realizing systems plays a key role in regulation of the stress reaction [12]. At the present time this model is commonly recognized. At the same time, the contribution of NO to the regulation of stress reaction, which may be very considerable, has remained unstudied.
Evaluating the effects of various stressors on NO production, different authors found both increases and decreases in NO (the table). Generally, increased NO production was observed under the action of short-term or moderate stressors while the decreased NO production was observed in the application of long-term and detrimental impacts of environmental factors [22]. Therefore, it can be suggested that the increased NO production corresponds to the stage of mobilization in response to the appropriate stress reaction while the decreased NO production corresponds to the stage of exhaustion in excessive stress reaction.
The stress-induced increase in NO synthesis can occur both due to activation of preexisting NO-synthase [23, 24] and at the expense of increased NO-synthase formation de novo [25, 26].
The activation of NO-synthase can be induced by: 1) increased concentration of intracellular calcium [27, 28]; 2) activation of free-radical oxidation [29]; and 3) increased concentration of free fatty acids [30].
The stress induction of the genes coding NO-synthase can occur as a result of activation of NFkB, a transcription factor. The NFkB activation can be induced by free radicals, calcium, cytokines, interleukins, and TNF-alpha [27, 31, 32].
It has been shown that levels of the factors activating or inducing NO-synthase are nonspecifically increased by any stress [1, 9, 33].
Besides, there exists a hypothesis that the increase in NO level may be independent of NO-synthase; NO may be formed from nitrites by nitrite reductase reactions, for instance, in hypoxia [34], or by a nonenzymic chemical transformation as demonstrated for ischemia of the isolated heart [35]. However activation of these mechanisms is related with the action of specific factors such as hypoxia or ischemia, rather than with the nonspecific stress reaction.
The right part of the scheme illustrates possible NO-dependent mechanisms of control to the stress reaction.
The structural, morphological basis of the NO-dependent control to central links of the stress reaction is provided by the fact that the pituitary gland receives an extensively ramified NO-ergic innervation from the hypothalamus [36]. NO modulates secretion of major pituitary stress hormones such as prolactin [37], luteinizing hormone [38], CRF [39], vasopressin [40], and growth hormone [41]. This suggests an important role of NO in regulation of the pituitary function and, consistently, in prevention of excessive activation of the stress system central division. This hypothesis is supported by the data obtained in our laboratory that the NO donor (dinitrosyl iron complex, DNIC) considerably reduces stress-related behavior of rats in open field tests.
When limiting the release of pituitary stress hormones, NO already by this mechanism reduces the activity of the peripheral division of the stress system. Nevertheless, there are also other NO-dependent mechanisms which limit the stress reaction directly at the level of its peripheral link.
It follows from the scheme (Fig. 1) that NO can block the peripheral discharge of stress hormones and directly protect cells and organs from stress damage.
NO restricts the release of sympathetic transmitters both at the level of the adrenals [42, 43] and at nerve terminals [44, 45]. NO-ergic neurons densely innervate the adrenal medulla. Furthermore, the nerve fibers directly contact with chromaffin cells which synthesize catecholamines [46]. Due to this fact, NO efficiently suppresses the stress discharge of catecholamines from the adrenal glands [42]. At the level of nerve terminals, NO suppresses catecholamine release from prejunctional membranes [44]. The process may be efficiency potentiated by NO that is released from the same sympathetic nerve terminals [47]. The important role of NO in limiting the peripheral release of catecholamines is supported by the data that inhibition of NO synthesis results in a pronounced activation of the sympathetic nervous system and stable hypertension [42, 48, 49].
Analyzing the NO-dependent mechanisms of local defense, it is reasonable to list once more the principal links in the stress damage: activation of free-radical oxidation, calcium overload of cells, protein denaturation, damage to cell membranes, and increased platelet aggregation and adhesion.
One protective mechanism of NO is related with its ability to activate antioxidant enzymes [50, 51] and expression of antioxidant-coding genes [5, 52]. In addition, the NO molecule itself possesses antioxidant properties [53]. These mechanisms may underlie the efficient limitation of free-radical activation in stress.
It has been shown that NO can prevent the increase in intracellular Ca2+ [54, 55]. This effect of NO can limit the detrimental effect of calcium on the heart and blood vessels. It is especially important that this effect takes place in conditions of Ca2+-ATPase inhibition [56] which is a situation typical for stress damage [57].
Recently, it has appeared that NO activates synthesis of protective stress proteins (Hsp70) [58, 59]. Hsp70 participate in renaturation of proteins injured by stress [60]. This means that the NO-dependent activation of Hsp70 can constitute an important mechanism of antistress cell defense.
One of the mechanisms of stress-induced damage to the membrane is related with prostaglandins of the E group and I2 [61]. It has been demonstrated that NO can activate cyclooxygenases and, thereby, stimulate synthesis of these cytoprotective prostaglandins [62, 63]. There are also some opposing data available that NO can inhibit cyclooxygenases by binding their hemoprotein group [64]. The mode of NO influence on prostaglandin synthesis is supposed to be determined by the duration of exposure and by the NO concentration [65].
Finally, the data that NO plays a key role in platelet aggregation and adhesion [66, 67] suggest that NO can limit the enhancement of throbogenesis induced by stress and, consistently, reduce such complications of long-term stress as ischemic damage to the heart and brain.
Therefore the presented data allow the hypothesis that NO contributes to the regulation of stress reaction through limitation of its over-activation and detrimental effects both at the central and peripheral levels.
If this hypothesis is correct, then an increase in the NO level caused, for example, by NO donors would limit the over-activation of the stress system, and, vice versa, a decrease in the NO production caused, for example, by NO-synthase inhibitors would potentiate detrimental effects of environmental factors. We have carried out respective verification experiments.
The NO production in the organism was evaluated using the EPR method [68] before and immediately after acute exposure to environmental factors (experiments were carried out together with V. D. Mikoyan, L. N. Kubrina, and A. F. Vanin, Institute of Chemical Physics, Moscow). We used dinitrosyl iron complex (DNIC) (200 µg/kg) (DNIC was a generous gift of Prof. A. F. Vanin, Institute of Chemical Physics, Moscow) as an NO donor. This NO donor has an important advantage over other known NO donors (nitroglycerin, sodium nitroprusside, SIN-1, SNAP, etc.): DNIC is a natural biological NO-containing compound which is formed as a result of the interaction between NO, iron, and SH-containing proteins [69]. To reduce the NO production we used Nomega-nitro-L-arginine (L-NNA) (50 mg/kg), an NO-synthase inhibitor.
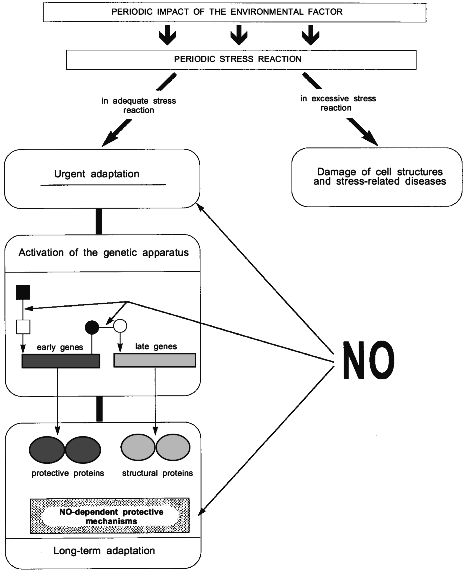
Figure 2 presents the data on changes in the NO production and on the effect of NO donor and NO-synthase inhibitor on the resistance of rats to four different potent stressors: 1) swimming with a heavy load until refusal of swimming; 2) immobilization with water immersion for 3 h resulting in pronounced stomach ulceration; 3) acute hypobaric hypoxia induced by an altitude chamber exposure to the simulated altitude of 11,000 m until termination of respiratory movements; and 4) acute heat shock for 30 min, which resulted in acute hypotension and death of some animals.
It is seen that, by the end of the session of exhausting swimming or long-term immobilization, NO generation was virtually completely suppressed. In these conditions, DNIC significantly potentiated while L-NNA considerably decreased both the duration of swimming and the area of erosive ulcers following immobilization. These results are in principle agreement with the hypothesis on the antistress role of NO.Fig. 2. Effect of the NO donor (DNIC) and the NOS inhibitor (L-NNA) on the resistance of rats to swimming with a load (a), restraint stress (b), acute hypoxia (c), and heat stroke (d): 1) control; 2) DNIC; 3) L-NNA. Ordinate: a) duration of swimming (min); b) total area of stomach mucosa ulcers (mm2); c) survival of animals in acute hypoxia (min); d) death rate (%).
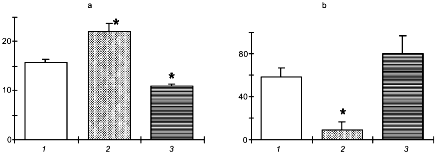
However, acute hypoxia and heat shock induced the NO over-production. In these conditions the NO-synthase inhibitor exerted a pronounced protective effect which was evident as improved survival of animals.
When analyzing the observed opposite effects of NO-synthase inhibitor on the animal's resistance to different stressors, one should keep in mind that the animal's response to the action of detrimental factors consists of nonspecific and specific stress reactions. In this process, NO not only plays a role in the stress reaction but can also be involved in mechanisms of specific damage. Depending on the nature of acting factor, the damage may be related with either hypo- or over-production of NO. For instance, restraint stress increases NO production in the brain structures responsible for the control of nonspecific stress reaction [25] and decreases the NO production in the stomach, leading to typical stress ulcers [70, 71].
NO hypo-production can be induced by low-density lipoproteins [72], high glucose concentrations [73], and ischemia [74]. The attenuation of NO synthesis increases the vascular tone and blood coagulation and compromises immunity, contributing thereby to the development of hypertension, atherosclerosis, diabetes, thrombosis, ischemic heart disease, infections, and tumor growth [75].
Excessive NO can induce injuries through inhibition of oxidative phosphorylation in mitochondria, DNA breakage, inhibition of enzymes in the tricarboxylic acid cycle, and formation of highly toxic peroxynitrites [31]. NO over-production causes excessive vasodilation and suppression of vasoconstriction, which constitute an important link in the pathogenesis of acute hypotension in heat [76, 77], cardiogenic [78], septic, and other types of shock [2, 75].
Therefore, the role of NO in an organism's responses to environmental factors is not so equivocal as it would have seemed at the first glance. This place is apparently determined by the relationship between the stress-limiting role of NO in the nonspecific stress reaction and the pathogenetic role of altered NO production in the specific damage under the action of a specific environmental factor.
This can explain the opposite effects of NO-synthase inhibitor on the organism's resistance to different stressors observed in our (Fig. 2) experiments and by other authors [79].
Probably, the NO-synthase inhibition attenuated the stress-limiting, that is, the protective function of NO in the nonspecific stress reaction. In the instances where the nonspecific detrimental effect of stressor was related with the suppressed NO production, the NO-synthase inhibitor decreased the organism's resistance to the stressor, and, vice versa, when the damage was mediated by NO over-production, the NO-synthase inhibitor enhanced the resistance.
On the whole the described data allow four principal statements on the role of NO in responses of the organism to stress can be elucidated.
1. Stress induced by diverse environmental factors is an activator of NO synthesis.
2. NO participates in the limitation of catecholamine release, a major component of the stress reaction.
3. NO itself or via the activation of local endogenous protective systems enhances the resistance of cells, organs, and the entire organism to detrimental impacts of the environment.
4. NO donors and NO-synthase inhibitors can provide a directed influence on stress responses of the organism.
The stress reaction is not an isolated phenomenon. From the very beginning, it occurs as a necessary link of adaptation to the environment. Correspondingly, one can believe that the systems of NO generation are involved not only in the control of stress responses but also in the formation of an organism's adaptation to environmental factors.
2. THE ROLE OF NO IN ADAPTATION
There are two major stages in the development of individual adaptation: urgent and long-term. Urgent and long-term adaptation are successive stages of the same process, adaptive increase in the organism's resistance to detrimental factors [1, 19]. Nevertheless, these stages involve different mechanisms and have different biological significance.
The scheme in Fig. 3 summarizes the principal stages of individual adaptation and demonstrates the possible role of NO in this process.
It is seen that the stress reaction can result in two alternative events: 1) excessive or long-term stress reaction can induce damage to cell structures and stress-related diseases, or 2) adequate stress reaction activates the mechanisms aimed at the protection of the organism against stress, i.e., at the development of the adaptive effect of stress reaction or, in other words, urgent adaptation.Fig. 3. The role of NO in formation of long-term adaptation to environmental factors (see text for explanations). Black square and circle, inactive transcription factors; light square and circle, active transcription factors.
Urgent adaptation is provided by pre-existing mechanisms. This stage involves activation of stress-limiting systems; mobilization of energy and structural resources and their transportation to the functional system responsible for the adaptation; rapid changes in activities of major lipid-dependent membrane proteins, enzymes, receptors and ion transport channels; increased synthesis of second messengers; and activation of cell metabolism [1, 12, 20, 21].
The mobilization of these mechanisms is followed by the stage of gene activation, which plays the key role in the transition from urgent to long-term adaptation.
Mechanisms of the genetic apparatus activation and the role of specific genes in adaptation can be characterized in many respects by the notion of a "black box". In general outline, the process of adaptation involves successive coordinated activation initially of early regulatory genes coding protooncogenes and stress proteins Hsp70, and then of late structural genes coding structural proteins [80] such as myosin, Ca2+-ATPase, antioxidant enzymes, etc.
The activation of early genes and the increase in Hsp70 and protooncogene synthesis constitute a nonspecific link in adaptation because they occur in adaptation to different factors [21].
Hsp70 were shown to be involved in the three most important nonspecific mechanisms of genetic provision for adaptation and its protective effects [21] (Fig. 3). The first mechanism is in that Hsp70 can perform a function of nuclear signals to activate the expression of late genes [80]. The second mechanism is related to the participation of Hsp70 in regulation of folding and intracellular transport of newly synthesized proteins [81]. This is especially important in the process of adaptation, when protein synthesis is increased. The third mechanism is related to protective properties of Hsp70 as such. Hsp70 can limit stress damage by disaggregating abnormal protein aggregates [60], participating in disposal of damaged proteins [82], and increasing the potency of antioxidant enzymes [83].
Subsequent activation of late structural genes is specific because the activated genes can provide adaptation to a specific factor. For instance, the genes coding the proteins involved in oxygen transport, growth of coronary blood vessels, and erythropoiesis [84, 85] are activated in adaptation to hypoxia; the genes coding thermogenin, lipoprotein lipase, and other proteins and enzymes which ensure enhanced heat production and growth of brown adipose tissue are activated in adaptation to cold [86], etc.
In the course of adaptation, the activation of the genetic apparatus provides a higher level of stress-limiting system activity [21, 87]. This results step by step in a virtually complete fading of the stress reaction in response to the next action of environmental factor in the adapted organism [88]. This is precisely the state that distinguishes long-term from urgent adaptation.
At the stage of urgent adaptation, the role of NO is defined primarily by the above-described participation of NO-dependent mechanism in the limitation of excessive stress reaction and its detrimental effects. In other words, at this stage, NO enhances the probability of the adaptive effect of stress reaction. In addition, NO dilates blood vessels in the organs responsible for adaptation and thereby provides redistribution of oxygen and substrates from non-active nerve centers, muscle groups and internal organs to the functional system performing the adaptive response.
A hypothesis on the role of NO in long-term adaptation was put forward several years ago, just after the discovery that adaptation to intermittent restraint stress and adaptation to intermittent exercise were accompanied by a pronounced increase in NO generation [89, 90]. Data about changes in NO production in adaptation to hypoxia are rather controversial [91-94]. However, it was shown that adaptation to hypoxia increased the expression of the NO-synthase gene [91] and the NO-synthase content [92]. These results suggest that in this type of adaptation, the absence of consistent changes in NO production may be an overall result of two simultaneous processes: increased NO synthesis and increased NO sequestration.
To obtain decisive evidence in favor of the suggestion that NO is indeed a necessary link in the development of long-term adaptation, we evaluated the effect of NO donor and NO-synthase inhibitor on the formation of protective effects of different types of adaptation and also elucidated the possibility of reproducing the protective effects of adaptation by a course of NO donor injections.
Experiments were carried out on male Wistar rats. The protective effect of adaptation to intermittent restraint stress, exercise, or hypoxia was evaluated respectively by the decrease in the area of stomach ulcers induced by restraint stress, by the increase in the duration of swimming with a load, and by the improved survival of animals exposed to the simulated altitude of 11,000 m above sea level.
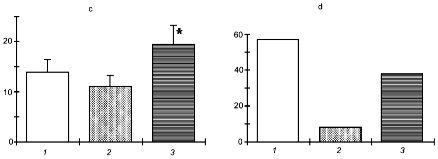
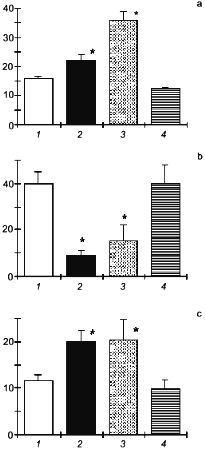
Figure 4 shows the results of these experiments (experiments were carried out together with N. P. Aimasheva, T. A. Zenina, E. B. Malenyuk, and L. Yu. Golubeva). It is seen that in all types of adaptation, the NO donor increased its efficiency while the NO-synthase inhibitor virtually completely prevented the development of the protective effect of adaptation. Therefore, using targeted modifications of the NO generation system one can control the adaptive process.
The use of exogenous NO donors is in essence an "imitation" of stress-limiting systems of the organism because the organism itself produces NO in the conditions of stress reaction. In our experiments, the NO donor as such, without adaptation, enhanced the organism's resistance to restraint stress, strenuous exercise, and acute hypoxia.Fig. 4. Effect of the NO donor (DNIC) and the NOS inhibitor (L-NNA) on the development of adaptation to exercise (a), restraint stress (b), and hypoxia (c): 1) control; 2) adaptation; 3) adaptation + DNIC; 4) adaptation + L-NNA. Ordinate: a) duration of swimming (min); b) total area of stomach mucosa ulcers (mm2); c) survival of animals in acute hypoxia (min).
Therefore, the presented data decisively show that NO plays a substantial role in the development of an organism's adaptation to environmental factors.
Importantly, irrespective of the type of adaptation, the NO-synthase inhibitor always prevented adaptation while the NO donor potentiated and reproduced the protective effects of adaptation. This indicates that NO is involved primarily in nonspecific mechanisms of adaptation.
As mentioned above, protooncogenes and Hsp70 play a key role in nonspecific effects of adaptation. There are presently only a little data on the effect of NO on protooncogene expression. At the same time, some data suggest that NO can either directly activate the genes [95] or potentiate their expression induced by Ca2+ [96]. The role of NO in the stress-induced activation of Hsp70 synthesis can be considered as proven. It was established that the NO-synthase inhibitor prevented the Hsp70 synthesis induced by heat shock [59], whereas the NO donor per se induced Hsp70 accumulation in cultured cells and in the whole organism [58, 97]. These data have led us to a hypothesis that the role of NO in nonspecific mechanisms of adaptation is related with the induction of Hsp70 synthesis. The simplest approach to verification of this hypothesis was to study the effect of NO-synthase inhibition on the accumulation of Hsp70 in the course of adaptation.
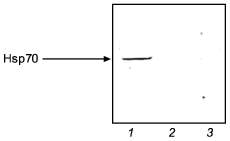
Figure 5 illustrates the effect of NO-synthase inhibitor (L-NNA) on the activation of Hsp70 synthesis in adaptation to intermittent hypoxia. It is seen that a course of mild hypoxic exposures induced a pronounced Hsp70 accumulation and enhanced the organism resistance to severe hypoxia. L-NNA completely prevented both the adaptive accumulation of Hsp70 and the development of adaptive defense.
The results suggest that, at least in adaptation to hypoxia, a NO-related mechanism occurs, which mediates activation of Hsp70 synthesis and provides the adaptational defense.Fig. 5. Effect of the NOS inhibitor (L-NNA) on Hsp70 accumulation in adaptation to hypoxia: 1) adaptation; 2) adaptation + L-NNA; 3) control.
Some protein products of early genes perform the function of transcription factor for late genes. The data of Nunoshiba et al. [5] demonstrate that NO activates such factors as exemplified by SoxR, the transcription factor of genes coding antioxidant enzymes.
Therefore the role of NO in the activation of genetic apparatus during the transition from urgent to long-term adaptation is apparently in that NO participates both in the activation of early genes and in the signal transmission from early to late genes (Fig. 3).
The stage of advanced long-term adaptation is characterized by potent protective effects and virtually complete absence of stress reaction in response to the next action of the environmental factor [1]. At this stage the NO level is stably increased. The biological significance of this phenomenon is in that the phenomenon is targeted at maintaining the organism's resistance to proceeding influence of the environmental factor, without the stress reaction.
The entire sequence of activating the mechanisms of NO synthesis and the role of NO at different stages of adaptation can be illustrated by an example of physical training, the best studied form of adaptation. During acute exercise, the increased blood flow velocity (shear stress) results in activation of endothelial constitutive NO-synthase [4, 23, 26]. The increased NO production results in blood vessel dilation which is especially pronounced in the heart and working skeletal muscles [98, 99]. This provides an adequate oxygen and substrate supply to principal organs involved in the urgent response to physical load. In subsequent exercise, the activation of pre-existing NO-synthase is supplemented by the increased expression of the NO-synthase gene [4, 26, 100, 101]. At this stage the potency of NO-producing systems enhances not only in the organs undergoing urgent adaptation, but also in other organs such as the liver, kidney, or brain [89]. The generalized activation of NO synthesis provided by the expression of NO-synthase gene reflects the transformation of the urgent to the long-term stage of adaptation. At the stage of long-term adaptation, a stable increase in NO production contributes to the enhancement of the organism's resistance not only to exercise, but also to other factors such as stress [12, 102], hypoxia [103], ischemia/reperfusion [104], thrombosis [105], and inflammation inducers [106].
Similar results were obtained in studying the role of NO in different stages of adaptation to hypoxia [91, 92, 107]. Furthermore, increased NO synthesis was found primarily in the lung, the organ playing the key role in this type of adaptation.
On the whole the data on the role of NO in adaptation presented in this section can be summarized as follows.
1. In different types of adaptation to environmental factors, the NO production consistently increases: initially due to the activation of pre-existing NO-synthases and subsequently also due to the increased expression of a NO-synthase gene.
2. NO activates early regulatory genes (hsp70 and protooncogenes) and transcription factors of some late structural genes participating in adaptation.
3. Exogenous NO donors promote while NO-synthase inhibitors prevent the formation of long-term adaptation.
4. The protective effect of adaptation to diverse factors can be reproduced using NO donors.
Taken together, these data lead to the following conclusion. NO contributes to the formation of three principal stages of long-term adaptation: 1) at the stage of urgent adaptation, NO restricts excessive stress reaction and provides targeted redistribution of energy resources to the organs performing the adaptive response; 2) at the transitional stage, NO activates early genes and contributes to the signal transmission from early to late genes; and 3) at the stage of advanced long-term adaptation, NO participates in maintenance of the enhanced resistance of the organism in the absence of stress reaction.
3. THE SYSTEM OF NO GENERATION IS A NEWLY RECOGNIZED
STRESS-LIMITING SYSTEM
The following properties of NO-generating systems manifest themselves in stress and adaptive responses of an organism: 1) stress-inducibility; 2) capability to limit the stress reaction; 3) capability to increase the resistance to environmental factors; 4) capability of NO to increase and of NO-synthase inhibitors to decrease the resistance to stress and adaptability of the organism; and 5) capability to increase its activity in the process of adaptation. It is easy to note that these features completely correspond to the criteria for stress-limiting systems postulated by Meerson [10, 12, 19] (see Section 1). This allows us to define the system of NO generation as a newly recognized stress-limiting system. In accordance with the conventional terminology, the system should be named the NO-ergic stress-limiting system.
An important peculiarity of the NO-ergic stress-limiting system is that large amounts of NO, the major metabolite of this system, are highly toxic. This means that NO would have been unable to realize its stress-limiting functions without a mechanism within the system structure, which would limit the over-activation of NO production. Indeed it was shown that the NO production which rapidly increased during heat shock returned to the initial level very soon [59].
It has been established that in the organism, the NO over-production is limited primarily by negative feedback [108] which is realized by several mechanisms.
1. Direct inactivation of NO-synthase by NO binding to the heme-containing moiety of the enzyme [109, 110].
2. Suppression of the NO-synthase gene expression by preventing the activation of NFkB, the transcription factor of inducible NO-synthase [111].
3. Prevention the expression of the NO-synthase gene by inhibition of NFkB binding to DNA [112].
4. Induction and stabilization of IkB-alpha, an endogenous NFkB inhibitor [113].
5. NO-dependent activation of antioxidant enzyme synthesis [5]. This can reduce the number of free radicals in the cell and, consistently, decrease NFkB activation.
6. NO-dependent activation of Hsp70 synthesis [58, 59]. Hsp70 in turn can suppress the expression of inducible NO-synthase through the decreased NFkB activation [114].
The negative feedback mechanism of limiting the NO over-production is valid for all known NO-synthase isoforms [109, 110, 112].
The negative feedback mechanism apparently underlies the ability of exogenous NO, endogenous NO, and prior adaptation to different factors to restrict NO over-production and, correspondingly, to prevent acute hypotension and toxic effects of excessive NO in endotoxemic shock [115], inflammation [116], heat shock [117, 118], acute myocardial infarction [102, 119], occlusion shock [120], and anaphylactic shock [121].
Another characteristic feature of the newly discovered stress-limiting system is that this system is universal, i.e., it limits the stress reaction and is involved in adaptation both at the central and peripheral levels.
We believe that this feature is related to unique properties of both the NO molecule itself and the NO-synthase. The matter is that NO is synthesized in the organism in the form of NO·. Further, in the course of chemical reactions, NO· can be transformed into NO- or NO+. Each NO form has its own cell targets and, correspondingly, contributes to different intracellular processes. For instance, it is known that the guanylate cyclase activation in the course of NO-dependent vasodilation and the nerve signal transmission are related with NO·, while the regulation of enzyme and transcription factor activities mediated by nitrosylation of protein structures is related to NO+ [122, 123].
Besides that, the NO molecule can exist in different electronic states; the universal nature of the NO-ergic stress-limiting system is determined by the fact that NO-synthase possesses six cofactors to control its activity. This is the most controllable enzyme known in biochemistry [46]. This fact suggests that the NO generation system is the most universal and sensitive sensor for intracellular changes induced by environmental factors.
4. POSSIBLE TRENDS OF FURTHER INVESTIGATIONS
Among basic aspects of the problem of NO in stress and adaptation, very promising is the role of NO-dependent mechanisms of control to the genetic apparatus in the development of adaptation to environmental factors. Although much data is currently available on the effect of NO donors on activities of different genes, information about the role of NO in adaptive changes in gene activity, except for hsp70, is virtually absent.
Another promising basic aspect may follow from the fact that NO-synthase synthesizes NO· while the primary role in the regulation of gene activity, the key link of adaptation, belongs to NO+. This means that the cell possesses a mechanism of control to the electronic state of NO molecule. The nature of this mechanism and the mode of its operation in adaptation to environmental factors remain virtually unknown.
Promising clinical trends in the problem of NO in stress and adaptation stem from the fact that the decreased NO production is typical for many diseases and pathological conditions such as atherosclerosis, ischemic heart disease, thrombosis, diabetes, hypertension, pyloric sphincter stenosis, etc., largely involving stress [2-4, 75]. At the same time, NO over-production which leads to a lowering of blood pressure and disorders of vasoconstrictor responses, is characteristic of shocks of different origin [2, 75, 76]. Application of adaptation methods targeted at increasing the NO production may prove an efficient means of prevention and treatment of diseases associated with deficiency or over-production of NO. The efficiency of such an approach has already been demonstrated in experiments [102, 103, 117, 119].
In addition, targeted modifications of the NO generation system may be also useful for improving the adaptive responses in sport, space, and aviation medicine.
The most promising pharmacological trends appear to be the following: 1) development and elucidation of new antistress drugs based on their capacity to influence consistently the NO generation, and 2) study of NO-dependent mechanisms in the action of known drugs possessing antistress activity (for example, adaptogens).
In developing methods of prophylaxis and treatment aimed at modulation of the NO generation system, the major task is to maintain or enhance the protective and physiological action of NO and at the same time to eliminate or restrict its detrimental effects.
The study was financed by the Russian Foundation for Basic Research (grant Nos. 97-04-48370, 97-04-48371, 96-15-97030).
REFERENCES
1.Meerson, F. Z. (1984) Adaptation, Stress and
Prophylaxis, Springer Verlag, Berlin.
2.Star, R. A. (1993) Am. J. Med. Sci.,
306, 348-358.
3.Moncada, S. (1994) J. Hypertension,
12, Suppl. 10, S35-S39.
4.Busse, R., and Fleming, I. (1995) Ann. Med.,
27, 331-340.
5.Nunoshiba, T., de Rojas-Walker, T., Wishnok, J. S.,
Tannenbaum, S. R., and Demple, B. (1993) Proc. Natl. Acad. Sci.
USA, 90, 9993-9997.
6.Shyy, Y.-J., Hsieh, H.-J., Usami, S., and Chien, S.
(1994) Proc. Natl. Acad. Sci. USA, 91, 4678-4682.
7.Malek, I. (1992) Am. J. Physiol.,
263, C389-C396.
8.Pantopoulos, K., Weiss, G., and Hentze, W. (1994)
Trends Cell Biol., 4, 82-86.
9.Stratakis, C. A., and Chrousos, G. P. (1996)
Ann. N. Y. Acad. Sci., 771, 1-18.
10.Meerson, F. Z., and Manukhina, E. B. (1984) in
Stress and Heart Disease (Beamish, R. E., Singal, P. K., and
Dhalla, N. S., eds.) Martinus Nijhoff Publishing, Winnipeg, pp.
422-435.
11.Chrousos, G. P., and Gold, P. W. (1992) J.
Amer. Med. Assoc., 267, 1244-1252.
12.Meerson, F. Z., and Pshennikova, M. G. (1988)
Adaptation to Stress Situations and Physical Loads [in Russian],
Meditsina, Moscow.
13.Chrousos, G. P. (1992) Endocrinol. Metab.
Clin. N. Am., 21, 833-858.
14.Calogero, A., Galluci, W. T., Gold, P. W., and
Chrousos, G. P. (1988) J. Clin. Invest., 82, 767-774.
15.Aghajanian, G. K., and van der Maelen, O. P.
(1982) Science, 215, 1394-1400.
16.Bagdy, G., Calogero, A. E., Murphy, D., and
Szemeredi, K. (1989) Endocrinology, 165, 2664-2669.
17.Calogero, A. E., Galluci, W. T., Chrousos, G. P.,
and Gold, W. P. (1988) Brain Res., 463, 28-36.
18.Meerson, F. Z. (1984) Pathogenesis and
Prevention of Stress and Ischemic Damages of the Heart [in
Russian], Meditsina, Moscow.
19.Meerson, F. Z. (1993) Concept of Long-Term
Adaptation [in Russian], Delo, Moscow.
20.Meerson, F. Z., Malyshev, I. Yu., Zamotrinsky, A.
V., and Kopylov, Yu. N. (1996) J. Mol. Cell Cardiol., 28,
835-843.
21.Meerson, F. Z., and Malyshev, I. Yu. (1993)
Phenomenon of Adaptive Stabilization of Structures and Protection of
the Heart [in Russian], Nauka, Moscow.
22.Busse, R., Mulsch, A., Fleming, I., and Hecker,
M. (1993) Circulation, 87, Suppl. V, V18-V25.
23.Oltman, C. L., Parker, J. L., and Laughlin, M. H.
(1995) J. Appl. Physiol., 79, 33-40.
24.Hiral, T., Visneeski, M. D., Kearns, K. J.,
Zelis, R., and Müsch, T. I. (1994) J. Appl. Physiol.,
77, 1288-1293.
25.Calza, L., Giardino, L., and Ceccatelli, S.
(1993) NeuroReport, 4, 627-630.
26.Shen, W. Q., Zhang, X. P., Zhao, G., Wolin, M.
S., Sessa, W., and Hintze, T. H. (1995) Med. Sci. Sports Exer.,
27, 1125-1134.
27.Busse, R., and Fleming, I. (1995) Ann.
Med., 27, 331-340.
28.Graier, W. F., Sturek, M., and Kukovetz, W. R.
(1994) Endothelium, 1, 223-236.
29.Mittal, C. K. (1993) Biochem. Biophys. Res.
Commun., 193, 126-132.
30.Ignarro, L. J. (1989) FASEB J., 3,
31-36.
31.Kröncke, K.-D., Fehsel, K., and
Kolb-Bachofen, V. (1995) Biol. Chem. Hoppe-Seyler, 376,
327-343.
32.Jordan, M. L., Rominski, B., Jaquinsgersti, A.,
Geller, J., and Hoffman, R. A. (1995) Surgery, 118,
138-146.
33.Kovacheva, S., and Ribarov, S. R. (1995)
Lung, 173, 255-263.
34.Reutov, V. P. (1995) Uspekhi Biol. Khim.,
35, 189-228.
35.Zweier, J. L., Wang, P., Samouilov, A., and
Kuppusami, P. (1995) Nature Medicine, 1, 804-809.
36.Vanhatalo, S., and Soinila, S. (1995) J. Chem.
Neuroanat., 8, 165-173.
37.Duvilanski, B. H., Zambruno, C., Seilicovich, A.,
Pisera, D., Lasaga, M., Diaz, M. del C., Belova, N., Rettori, V., and
McCann, S. M. (1995) Proc. Natl. Acad. Sci. USA, 92,
170-174.
38.Ceccatelli, S., Hulting, A.-L., Zhang, X.,
Gustavsson, L., Villar, M., and Hokfelt, T. (1993) Proc. Natl. Acad.
Sci. USA, 90, 11292-11296.
39.Karanth, S., Lyson, K., and McCann, S. N. (1993)
Proc. Natl. Acad. Sci. USA, 90, 3383-3387.
40.Ota, N., Crofton, J. T., Festavan, G. H., and
Share, L. (1993) Amer. J. Physiol., 57, 955-959.
41.Kato, N. (1992) Endocrinology, 131,
2133-2138.
42.Zanchi, A., Shaad, N. C., Osterheld, M. C.,
Grouzmann, E., Nussberger, J., Brunner, H. R., and Waeber, B. (1995)
Amer. J. Physiol., 37, H2267-H2273.
43.Torres, M., Ceballos, G., and Rubio, R. (1994)
J. Neurochem., 63, 988-996.
44.Addicks, K., Bloch, W., and Feelisch, M. (1994)
Microscopy Res. Tech., 29, 161-168.
45.Schwarz, P., Diem, R., Dun, N. J., and
Forstermann, U. (1995) Circ. Res., 77, 841-848.
46.Snyder, S. H., and Bredt, D. S. (1991)
TIPS, 12, 125-128.
47.Toda, N., Yoshida, K., and Okamura, T. (1991)
Naunyn-Schmiedeberg's Arch. Pharmacol., 343, 221-224.
48.Sander, M., Hansen, P. G., and Victor, R. G.
(1995) Hypertension, 26, 691-695.
49.Goodson, A. R., Leibold, J. M., and Gutterman, D.
D. (1994) Amer. J. Physiol., 36, H1272-H1278.
50.Dobashi, K., Pahan, K., Chahal, A., and Singh, I.
(1997) J. Neurochem., 68, 1806-1903.
51.Rotzinger, S., Aragon, C. M. G., Rogan, F., Amir,
S., and Amit, Z. (1995) Life Sci., 56, 1321-1324.
52.White, A. C., Maloney, E. K., Boostani, M. R.,
Hassoun, P. M., and Fanburg, B. L. (1995) Amer. J. Resp. Cell Mol.
Biol., 13, 442-448.
53.Kanner, J., Harel, S., and Granit, R. (1991)
Arch. Biochem. Biophys., 289, 130-136.
54.Minowa, T., Miwa, S., Kobayashi, S., Enoki, T.,
Zhang, X. F., Komuro, T., Iwamuro, Y., and Masaki, T. (1997) Brit.
J. Pharmacol., 120, 1536-1544.
55.Andriantsitohaina, R., Lagaud, G. J., Andre, A.,
Muller, B., and Stoclet, J. C. (1995) Amer. J. Physiol.,
268, H1223-H1231.
56.Petkov, G. V., and Boev, K. K. (1996)
Pflugers-Arch., 431, 928-935.
57.Meerson, F. Z., Arkhipenko, Yu. V., Rozhitskaya,
I. I., and Kagan, V. E. (1981) Byul. Eksp. Biol. Med., 9,
281-283.
58.Malyshev, I. Yu., Malugin, A. V., Golubeva, L.
Yu., Zenina, T. A., Manukhina, E. B., Mikoyan, V. D., and Vanin, A. F.
(1996) FEBS Lett., 391, 21-23.
59.Malyshev, I. Yu., Manukhina, E. B., Mikoyan, V.
D., Kubrina, L. N., and Vanin, A. F. (1995) FEBS Lett.,
370, 159-162.
60.Pelham, H. R. B. (1986) Cell, 46,
959-961.
61.Van Bilsen, M., Engels, W., van der Vusse, G. J.,
and Reneman, R. S. (1989) Mol. Cell Biochem., 88,
113-121.
62.Uno, H., Arakawa, T., Fukuda, T., Fujiwara, H. Y.
Y., Higuchi, K., Inoue, M., and Kobayashi, K. (1997)
Prostaglandins, 53, 153-162.
63.Salvemini, D., Misko, T. P., Masferrer, J. L.,
Seibert, K., Currie, M. G., and Needleman, P. (1993) Proc. Natl.
Acad. Sci. USA, 90, 7240-7244.
64.Kanner, J., Harel, S., and Granit, R. (1992)
Lipids, 27, 46-49.
65.Swierkosz, T. A., Mitchell, J. A., and Tomlinson,
T. A. (1994) Pol. J. Pharmacol., 46, 587-592.
66.Vallance, P., Benjamin, N., and Collier, J.
(1992) Eur. J. Clin. Pharmacol., 42, 37-41.
67.Radomski, M. W., Palmer, R. M., and Moncada, S.
(1987) Lancet, 2, 1057-1058.
68.Vanin, A. F., Mordvintcev, P. I., and Kleschev,
A. L. (1984) Stud. Biophys., 107, 135-142.
69.Vanin, A. F., Men’shikov, G. B., Moroz, I.
A., Mordvintcev, P. I., Serezhenkov, V. A., and Burbaev, D. S. (1992)
Biochim. Biophys. Acta, 1135, 275-279.
70.Whittle, B. J. R., Lopez-Bemonte, J., and
Moncada, S. (1990) Br. J. Pharmacol., 99, 607-611.
71.Ogino, K., Takeyama, Y., Ischiyama, H.,
Kobayashi, H., and Hobara, T. (1994) Pathophysiology, 1
(Suppl.), 391.
72.Keaney, J. F., and Vita, J. A. (1995) Progr.
Cardiovasc. Dis., 38, 129-154.
73.Bucala, R., Traccy, K. J., and Cerani, A. (1991)
J. Clin. Invest., 87, 432-438.
74.Cooke, J. P., and Tsao, P. S. (1993)
Circulation, 88, 2451-2454.
75.Pörsti, I., and Paakkari, I. (1995) Ann.
Med., 27, 407-420.
76.Manukhina, E. B., Malyshev, I. Yu., Mikoyan, V.
D., Kubrina, L. N., and Vanin, A. F. (1996) Byul. Eksp. Biol.
Med., 121, 520-523.
77.Manukhina, E. B., Azamatov, Z. Z., and Malyshev,
I. Yu. (1996) Byul. Eksp. Biol. Med., 122, 148-151.
78.Vanin, A. F., Lapshin, A. V., Manukhina, E. B.,
and Meerson, F. Z. (1994) in The Biology of Nitric Oxide. Pt. 3.
Clinical and Physiological Aspects (Moncada, S., Feelish, M.,
Busse, R., and Higgs, E. A., eds.) Portland Press, London, pp.
96-99.
79.Lopez-Belmonte, J., Whittle, B. J. R., and
Moncada, S. (1993) Br. J. Pharmacol., 108, 73-78.
80.Goelet, Ph. (1986) Nature, 322,
419-422.
81.Beckman, R., Mizzen, L., and Welch, W. (1990)
Science, 248, 850-854.
82.Hershko, A. (1988) J. Biol. Chem.,
263, 15237-15240.
83.Privalle, C. T., and Fridovich, I. (1987)
Proc. Natl. Acad. Sci. USA, 84, 2723-2726.
84.Ostadal, B., Kolar, F., Pelouch, V., Bass, A.,
Samunek, M., and Prochazka, J. (1989) Biomed. Biochem. Acta,
48, 558-562.
85.Scholz, H., Schurec, H. J., Eckardt, K. U., and
Bauer, C. (1990) Experientia, 46, 1197-1201.
86.Obregon, M. J., Jacobsson, A., Kirchgessner, T.,
Schotz, M. C., Cannon, B., and Nedergaard, J. (1989) Biochem.
J., 259, 341-346.
87.Meerson, F. Z. (1991) Adaptive Protection of
the Heart: Protecting Against Stress and Ischemic Damage, CRC
Press, Boca Raton.
88.Keim, K. L., and Sigg, E. B. (1978) Pharmacol.
Biochem. Behav., 4, 289-297.
89.Manukhina, E. B., Lapshin, A. V., Meerson, F. Z.,
Mikoyan, V. D., Kubrina, L. N., and Vanin, A. F. (1996) Fiziol. Zh.
im. I. M. Sechenova, 82, 54-60.
90.Meerson, F. Z., Mordvintcev, P. I., Manukhina, E.
B., Lapshin, A. V., Mikoyan, V. D., Kubrina, L. N., and Vanin, A. F.
(1994) in The Biology of Nitric Oxide. Pt. 3. Clinical and
Physiological Aspects (Moncada, S., Feelish, M., Busse, R., and
Higgs, E. A., eds.) Portland Press, London, pp. 182-185.
91.Shaul, P. W., North, A. J., Brannon, T. S.,
Ujiie, K., Wells, L. B., Nisen, P. A., Lowenstein, C. J., Snyder, S.
H., and Star, R. A. (1995) Am. J. Resp. Cell Mol. Biol.,
13, 167-174.
92.Xue, C., Rengasamy, A., Lecras, T. D., Koberna,
P. A., Dailey, G. C., and Johns, R. A. (1994) Am. J. Physiol.,
11, L667-L678.
93.Eichinger, M. R., and Walker, B. R. (1994) Am.
J. Physiol., 36, H2413-H2419.
94.Lapshin, A. V., Manukhina, E. B., Meerson, F. Z.,
Mikoyan, V. D., Kubrina, L. N., and Vanin, A. F. (1995) Hypoxia Med.
J., No. 1, 3-5.
95.Tassorelli, C., and Joseph, S. A. (1995) Brain
Res., 695, 37-44.
96.Peunova, N., and Enikolopov, G. (1993)
Nature, 364, 450-453.
97.Kim, Y.-M., de Vera, M. E., Watkins, S. C., and
Billiar, T. R. (1997) J. Biol. Chem., 272, 1402-1411.
98.Sun, D., Huang, A., Koller, A., and Kaley, G.
(1994) J. Appl. Physiol., 76, 2241-2247.
99.Dustingm, G. J. (1996) in Myocardial Ischemia:
Mechanisms, Reperfusion, Protection (Karmazyn, M., ed.) Birkhauser
Verlag, Basel, pp. 33-55.
100.Sessa, W. C., Pritchard, K., Seyedi, N., Wang,
J., and Hintze, T. H. (1994) Circ. Res., 74, 349-353.
101.Laughlin, M. H. (1995) Med. Sci. Sports
Exer., 27, 1135-1144.
102.Manukhina, E. B., Lapshin, A. V., and Meerson,
F. Z. (1996) Physiol. Res., 45, 261-266.
103.Aymasheva, N. P., Zenina, T. A., Manukhina, E.
B., Mikoyan, V. D., Kubrina, L. N., Vanin, A. F., and Malyshev, I. Yu.
(1997) Izvestiya RAN. Ser. Biol., No. 5, 652-656.
104.Wang, J., Wolin, M. S., and Hintze, T. H.
(1993) Circ. Res., 73, 829-838.
105.Wang, J. S., Jen, C. J., and Chen, H. I. (1995)
Atherosclerosis, Thrombosis and Vascular Biology, 15,
1668-1674.
106.Conn, C. A., Kozak, W. E., Tooten, P. C. J.,
Gruys, E., Borer, K. T., and Kluger, M. J. (1995) J. Appl.
Physiol., 78, 466-477.
107.Hampl, V., Cornfield, D. N., Cowan, N. J., and
Archer, S. L. (1995) Eur. Resp. J., 8, 515-522.
108.Assreuy, J., Cunha, F. Q., Liew, F. Y., and
Moncada, S. (1993) Br. J. Pharmacol., 108, 833-837.
109.Abusoud, H. M., Wang, J. L., Rousseau, D. L.,
Fukuto, J. M., Ignarro, L. J., and Stuehr, D. J. (1995) J. Biol.
Chem., 270, 22997-23006.
110.Ravichandran, L. V., Johns, R. A., and
Rangasamy, A. (1995) Amer. J. Physiol., 37,
H2216-H2223.
111.Colasanti, M., Persichini, T., Menegazzi, M.,
Mariotto, S., Giordano, E., Caldarera, C. M., Sogos, V., Lauro, G. M.,
and Suzuki, H. (1995) J. Biol. Chem., 270,
26731-26733.
112.Park, S. K., Lin, H. L., and Murphy, S. (1997)
Biochem. J., 322, 609-613.
113.Peng, H. B., Libby, P., and Liao, J. K. (1995)
J. Biol. Chem., 270, 14214-14219.
114.Feinstein, D. L., Galea, E., Aquino, D., Li, G.
C., Xu, H., and Reis, D. (1996) J. Biol. Chem., 271,
17724-17732.
115.Kumins, N. H., Hunt, J., Gamelli, R. L., and
Filkins, J. P. (1997) Shock, 7, 200-205.
116.Decaterina, R., Libby, P., Peng, H. B.,
Thunnickal, V. J., Rajavashisth, T. B., Gimbrone, M. A., Shin, W. S.,
and Liao, J. K. (1995) J. Clin. Invest., 95, 60-68.
117.Manukhina, E. B., Pokidyshev, D. A., and
Malyshev, I. Yu. (1997) Byul. Eksp. Biol. Med., 124,
380-383.
118.Manukhina, E. B., Pokidyshev, D. A., Malenyuk,
E. B., Malyshev, I. Yu., and Vanin, A. F. (1997) Izvestiya RAN. Ser.
Biol., No. 1, 54-58.
119.Meerson, F. Z., Manukhina, E. B., and Lapshin,
A. V. (1991) Biomed. Sci., 2, 623-628.
120.Aoki, N., Johnson, G., and Lefer, A. M. (1990)
Am. J. Physiol., 258, G275-G281.
121.Markovic, B. M., Dordevic, I. L. J., Lazarevic,
M., Sporcic, Z., Duric, V. J., and Jankovic, B. D. (1990) Period.
Biol., 92, 69.
122.Arnelle, D. R., and Stamler, J. S. (1995)
Arch. Biochem. Biophys., 318, 279-285.
123.Stamler, J. S., Singel, D. J., and Loscalzo, J.
(1992) Science, 258, 1898-1902.
124.Solodkov, A. P., and Bozhko, A. P. (1994)
Byul. Eksp. Biol. Med., 9, 245-249.
125.Mcquillan, I. P., Leung, G. K., Marsden, P. A.,
Kostyk, S. K., and Kourembanas, S. (1994) Am. J. Physiol.,
36, H1921-1927.
126.Trolin, G., Anden, T., and Hedenstrierna, G.
(1994) Acta Physiol. Scand., 151, 159-163.
127.Joannides, R., Haefeli, W. E., Linder, L.,
Richard, V., Bakkali, E. I., Hassan, H., Thuillez, C., and
Lüscher, T. (1995) Circulation, 91, 1314-1319.
128.Wilson, J. R., and Kapoor, S. (1993) J.
Appl. Physiol., 75, 2740-2744.
129.Hiral, T., Visneeski, M. D., Kearns, K. J.,
Zelis, R., and Musch, T. I. (1994) J. Appl. Physiol., 77,
1288-1293.
130.Endo, T., Imaizumi, T., Tagawa, T., Shiramoto,
M., Ando, S., and Takeshita, A. (1994) Circulation, 90,
2886-2890.
131.Carter, E. A., Derojaswalker, J., Tamir, S.,
Tannenbaum, Yu. Y. M., and Tompkins, R. G. (1994) Biochem. J.,
304, Pt. 1, 201-204.
132.Hall, D. M., Buettner, G. R., Matthes, R. D.,
and Gisolfi, C. V. (1994) J. Appl. Physiol., 77,
548-553.
133.Lubbe, A. S. (1994) Shock, 2,
179-184.
134.Kuroshima, A., Nagashima, T., and Ohinata, H.
(1995) in Abst. IV World Congr. of Adaptive Medicine,
Postgraduate Institute of Medical Education, Chandigarh, India, p.
113.
135.Kader, A., Frazzini, V. I., Baker, C. J.,
Solomon, R. A., and Trifiletti, R. R. (1994) Neurosurgery,
35, 272-277.
136.Castellani, S., Ungar, A., Lacava, G., Cantini,
C., Stefanile, C., Camaiti, A., Messeri, G., Coppo, M., Vallitti, B.,
Diserio, C., Brocchi, A., and Masotti, G. (1997) J. Lab. Clin.
Med., 129, 642-649.
137.Dietz, N. M., Rivers, J. M., Effener, S. E.,
Fix, R. T., Warner, D. O., and Joyner, M. J. (1994) J. Physiol.
(London), 480, Pt. 2, 361-368.
138.Persoons, J. H. A., Schornagel, K., Breve, J.,
Berkenbosch, F., and Kraal, G. (1995) Am. J. Resp. Crit. Care
Med., 152, 619-624.
139.Manukhina, E. B., Lapshin, A. V., Ustinova, E.
E., and Meerson, F. Z. (1989) Fiziol. Zh. SSSR im. I. M.
Sechenova, 75, 1409-1416.
140.Duval, D. L., Sieg, D. J., and Billings, R. E.
(1995) Arch. Biochem. Biophys., 316, 699-706.
141.Vile, G. F., Tanewiliitschew, A., and Tyrrell,
R. M. (1995) Photochem. Photobiol., 62, 463-468.
142.Gourine, A. V. (1994) in Thermal Balance in
Health and Disease: Recent Basic Research and Clinical Progress
(Zeisberger, Z., Schonbaum, E., and Lomax, P., eds.) Birkhauser Verlag
AG, Berlin, pp. 491-495.
143.Gilligan, D. M., Panza, J. A., Kilcoyne, C. M.,
Waclawiw, M. A., Casino, P. R., and Quyyumi, A. A. (1994)
Circulation, 90, 2853-2858.
144.Green, D. J., Cable, N. T., Fox, C., Rankin, J.
M., and Taylor, R. R. (1994) J. Appl. Physiol., 77,
1829-1833.
145.Komiyama, Y., Kimura, Y., Nishimura, N., Hara,
K., Mori, T., Okuda, K., Munakata, M., Masuda, M., Murakami, T., and
Takahashi, H. (1997) Clin. Exp. Hypertension, 19,
363-372.
146.Koller, A., Huang, A., Sun, D., and Kaley, G.
(1995) Circ. Res., 76, 544-550.
147.Chen, H., Li, H.-T., and Chen, C.-C. (1994)
Circulation, 90, 970-975.
148.Jansakui, C. (1995) Brit. J. Pharmacol.,
115, 587-594.