REVIEW: Nitric Oxide and Oxygen Radicals in Infection, Inflammation, and Cancer
H. Maeda* and T. Akaike
Department of Microbiology, Kumamoto University School of Medicine, 2-2-1 Honjo, Kumamoto 860-0811, Japan; fax: (+81) 96-362-8362; E-mail: msmaedah@gpo.kumamoto-u.ac.jp* To whom correspondence should be addressed.
Received March 24, 1998
In recent years, accumulated evidence indicates that free radical species and nitric oxide (NO) or its derivatives are the key denominators in carcinogenesis. Our present topics discussed in this article will focus on the biological significance of free radical generation induced by viral and bacterial infections. In influenza virus infection in mice, the level of xanthine oxidase (XO) at the infected sites was elevated to a great extent. The timing of paralleled induction of XO with that of inducible NO synthase (iNOS) indicates efficient simultaneous reaction: NO + O2·- --> ONOO- (peroxynitrite). Peroxynitrite formation was identified by immunostaining of nitrotyrosine at the local site of infected organs. Peroxynitrite exhibits unique chemical reactivities such as protein nitration, DNA-strand breakage, guanine nitration, etc., which may then bring about not only cytotoxic effect but also mutagenesis. Numbers of evidence in vitro and in vivo show that treatment with chemical carcinogens such as carbon tetrachloride and heterocyclic amines also generated superoxide. The chronic inflammatory reactions, e.g., zymosan- and silica-induced granuloma, revealed very similar free radical generation in vivo. In addition, most experimental solid tumors have elevated levels of iNOS in the tumor tissue, and NO thus generated facilitates vascular permeability, which accelerates nutritional supply to the tumor tissue and hence sustains the rapid tumor growth. These circumstantial evidences suggest that inflammatory responses induced by various pathogens would accelerate mutagenesis as well as tissue damage, whereas NO also sustains more effectively solid tumor growth when normal cells are transformed to tumor or carcinoma cells by the host-derived free radical species.
KEY WORDS: nitric oxide, superoxide, oxygen free radicals, peroxynitrite, nitric oxide synthase, xanthine oxidase, carcinogenesis, mutagenesis, infection, inflammation, cancer
Abbreviations: XO) xanthine oxidase; iNOS) inducible NO synthase; LPS) lipopolysaccharide; 8OHG) 8-OH-guanine; ONOO-) peroxynitrite; ADA) adenosine deaminase; BALF) bronchoalveolar lavage fluid; SOD) superoxide dismutase; CMV) cytomegalovirus; HIV-1) human immunodeficiency virus type 1; IFN-gamma) gamma-interferon; TNF-alpha) tumor necrosis factor alpha; L-NMMA) NG-monomethyl-L-arginine; PTIO) 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide; VPF) vascular permeability factor; VEGF) vascular endothelial growth factor; L-NAME) Nomega-nitro-L-arginine methyl ester.
A classic theory of the cause of cancer includes electromagnetic
radiation such as X-ray, chemicals such as heterocyclic amines, and
viral infections. There appears to be no common denominator among them.
In recent years, ample evidence showed that free radical species and
nitric oxide (NO) or its derivatives are the key denominators in
carcinogenesis. Oxygen radicals and nitrogen oxide derivatives can
effectively damage DNA and hence causing mutations, and probably they
are involved in multiple steps of carcinogenesis in vivo, which
usually requiring a long time of exposure. Inflammatory tissues are now
well known to have activated state of generation of superoxide
(O2·-) and NO. All
these individual event can be unified based on the biological or
biochemical action of the oxygen free radicals and NO derivatives (Fig.
1).
In this review, we present some of our data showing that NO, superoxide, and their reaction product peroxynitrite generated in infectious diseases have a great consequence in the progression and pathogenesis of diseases; namely they become mediators of inflammation, modify proteins and damage nucleic acids. These events involving chronic inflammation for 10 or more years may trigger carcinogenesis, while the long term process called "progression" evolves to become genuine cancer in vivo.Fig. 1. A central theme and universal mechanism in carcinogenesis. Interaction of free radicals and DNA in infection, inflammation, and carcinogens, which leads to cancer.
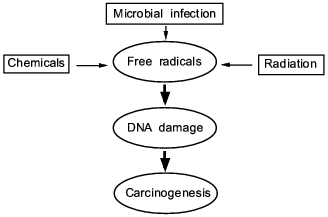
FREE RADICAL GENERATION AND ITS BIOLOGICAL RELEVANCE IN
CARCINOGENESIS CAUSED BY INFECTIONS
Proinflammatory cytokines such as interferon-gamma, interleukin-1beta, and other cytokines are modulators of the inflammatory reactions and many of them facilitate induction of the inducible isoform of NO synthase (iNOS), thus they could mediate excessive production of NO [1]. Among the reactive molecular species which might injure nucleic acids, we believe superoxide and NO are the most important molecular species; the former is generated by such enzymes as xanthine oxidase (XO) and NADPH oxidase, and the latter by NOS. Hydrogen peroxide (H2O2) is also important if not converted into H2O and O2 by catalase. Namely, H2O2 can be readily converted to more reactive hydroxyl radical (·OH) via the Fenton reaction in the presence of transition metals, which is likely to occur in biological systems. Biosynthesis of NO as well as superoxide generation are expressed in defense-oriented white blood cells such as macrophage and polymorphonuclear cells, endothelial cells, and other tissue cells. These cells are activated, or these enzymes are induced to produce free radical species most commonly in microbial infections and by other stress or insults to the hosts, such as X-ray radiation or chemical carcinogens. Two common mechanisms of NOS induction or activation are: 1) by lipopolysaccharide (LPS) and teichoic acid of gram-negative and -positive bacteria [1-3], or 2) bradykinin which very frequently takes place upon bacterial infection [4, 5]. Well-studied events along this line include ischemia followed by reperfusion or embolization. Once these reactive molecular species (or free radicals) are generated during the host's inflammatory responses against microbial pathogens, they can induce damages to the host's DNA, enzymes/proteins, and other vital molecules. Some oxygen radicals such as hydroxyl radical, which is highly reactive and short-lived, may be converted into more stable (having longer half-life) lipid peroxide radicals (LOO·), which travel long distances in vivo and might accumulate in the fat rich organs.
Accordingly, infections might become the cause of cancer in theory. Then, one might ask if there really are such cases that microbial infections can cause cancer. At least five examples of epidemiological study shown with asterisks in the table have more solid evidences suggesting the link between free radicals formation in the tissue and carcinogenesis. The recognition of the importance of free radicals in carcinogenesis is recently increasing [6-8], because evidences of extensive free radical formation in the pathogenesis of infections and inflammation convince us more than before the cause and effect relationship of infections and cancers.
Cancer caused by various infections

Although the free radical theory has played a rather minor role in the previous study of cancer etiology [6-8], it appears more realistic in view of more recent findings of generation of superoxide, peroxynitrite, and NO in infectious diseases. It is also intriguing that these reactive molecular species are found to be generated upon treatment of animals with more common chemicals, not to mention benzopyrene or heterocyclic amines.
Gastric cancer is the second commonest cancer in Japan, and it now appears to be caused by a strange bacterium called Helicobacter pylori. H. pylori can produce a chronic infection manifested frequently as gastric and gastroduodenal ulcer for a period longer than ten years. Ohshima et al. in Lyon, France, showed that DNA nitration of guanine and formation of 8-OH-guanine (8OHG) do exist in H. pylori infected human gastric mucosa [9]. Kawanishi et al. in Kyoto, Japan, and Ohshima's group also reported that peroxynitrite can damage nucleic acids [10, 11]. In the human gastric mucosa with chronic H. pylori infection, pronounced DNA damage, particularly formation of 8OHG, is reported by Baik et al. very recently [12], which is additional in vivo evidence that infections cause DNA damage. In case of chronic typhoid and paratyphoid carriers infected with Salmonella typhi and S. paratyphi A, B, the incidence of the cancers of the gallbladder, pancreas, etc. are also reported to be high. This again may provide another example of cancer incidence involving bacterial infection [13].
Moreover, various microbial infections including hepatitis B and C virus, Epstein--Barr (EB) virus, and others including parasites (Opisthorchis viverrini and Schistosoma mansoni) have been suggested to be risk factors of hepatoma, gastric cancer, and other types of cancer (table), and the cause may be closely related to the formation of free radicals and reactive nitrogen oxides, particularly peroxynitrite (ONOO-) [14].
VIRUS INFECTIONS AND OXYGEN FREE RADICAL GENERATION
Human hepatoma is most frequently caused by hepatitis B and C virus. The etiology of gastric cancer is also reported to involve EB virus [15]. These viruses are known to induce chronic or persistent infections. Now we will describe some evidence that viral infection results in generation of free radicals, although the model presented here is that of acute-semiacute infection.
One example is a case of influenza virus infections in mice in which the actual cause of death of infected animal is not the virus per se, but we found free radicals are more responsible [16-20].
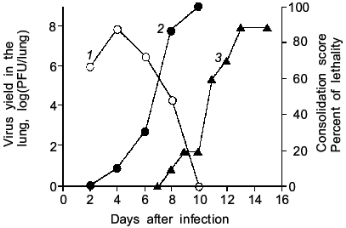
As illustrated in Fig. 2, the mice infected with influenza virus started to die on day 8 or later and almost all died of severe pneumonia on day 14. However, virus titer in the lung became undetectable by the plaque-forming assay on day 10 or later. Apparently, no infectious virus was found at least in the primary site of the virus infection at the time point of maximal death of mice. Then, one will question what caused the death of the mice.
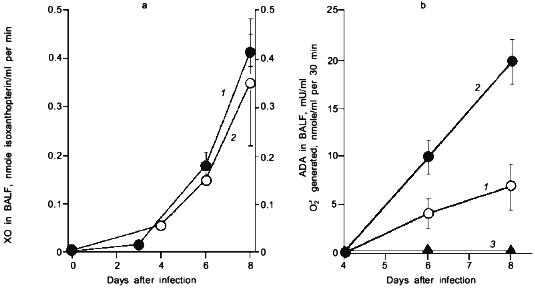
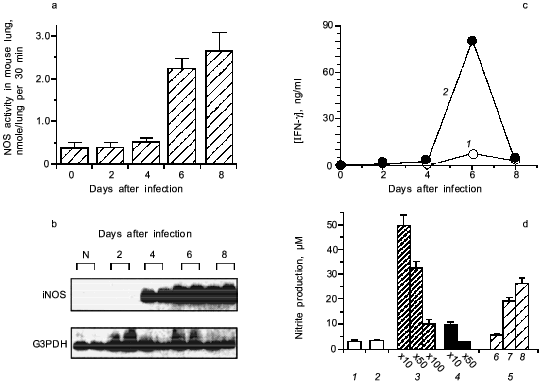
When we quantified the amount of adenosine deaminase (ADA) and XO in the bronchoalveolar lavage fluid (BALF) of the infected mice, these values were greatly increased as shown in Fig. 3a. Several hundred-fold increase in XO activity in the BALF of infected mice was observed. The enhanced capacity to generate superoxide in BALF is shown in Fig. 3b. With the addition of adenosine to the BALF obtained from the infected mice, a greater increase in superoxide generation was observed as the time after infection progresses (Fig. 3b).Fig. 2. Time course of viral replication (1), score of consolidation in the lung (2), and survival rate (3) of influenza virus-infected mice. Mice were inoculated by inhalation with the influenza virus A/Kumamoto/Y5/67(H2N2) at twice the LD50 dose [17].
To conclude these findings, the infection of influenza virus was coupled with induction of ADA and XO and generation of superoxide, in which catabolic degradation of nucleic acids to adenosine facilitates superoxide generation as a fuel for XO, particularly to hypoxanthine and xanthine productions in this case [17].Fig. 3. a) Induction of xanthine oxidase (XO) (1) and adenosine deaminase (ADA) (2) activities in the bronchoalveolar lavage fluid (BALF) of the mice infected with influenza virus. b) O2·- generation in supernatant of BALF in the absence (1) or presence of adenosine (2) or allopurine (3). O2·- generated was determined spectroscopically on the basis of SOD-inhibitable reduction of cytochrome c. Data are means ± SEM (n = 3) [17].
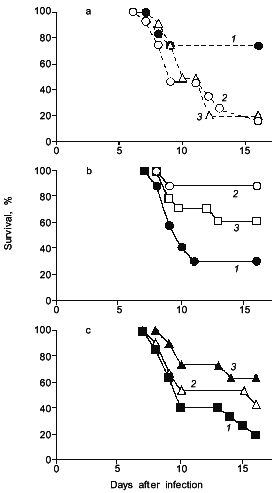
Interestingly, when adenosine was injected to the influenza infected mice, mice died more quickly and more extensively (Fig. 4a) [19]. It was also demonstrated as shown in Fig. 4 (b and c) that scavenging of superoxide by administration of superoxide dismutase (SOD), which is conjugated with biocompatible polymer (pyran copolymer) to improve pharmacokinetics or plasma retention time, or by injecting an inhibitor of XO such as allopurinol which suppresses the superoxide production, indeed improved the survival rate of mice [16, 17], thus superoxide toxicity hypothesis seems valid.
Similar examples of superoxide generation are now described for other virus infections [18-20]. More specifically, it is reported by Ikeda et al. that XO activity in the mouse lung was elevated appreciably after cytomegalovirus (CMV) infection in mice, and the number of pulmonary lesions significantly ameliorated after treatment with SOD or allopurinol [21]. The effect of recombinant human Mn-SOD was examined in mice infected with influenza virus (A or B) by Sidwell et al., who found a beneficial effect of the SOD treatment, particularly on the pulmonary function of the virus-infected animals [22].Fig. 4. The effects of adenosine, SOD, and allopurinol on influenza virus-infected mice. a) Adenosine was given intraperitoneally ((1) is control; 0.5 (2), 0.1 mg (3)) daily, from days 4 to 7. b) SOD (3) or pyran--SOD (conjugate of pyran copolymer with Cu,Zn-SOD) (2) were intravenously administered (200 (2), 1000 U (3)) daily, from days 5 to 8 ((1) is control, vehicle alone). c) Allopurinol (1 (2), 2 mg (3)) was orally administered daily, from days 4 to 7 ((1) is control, vehicle alone). In these experiments, each group contains 10 to 15 mice [17, 19].
One interesting observation is that latent infection with human immunodeficiency virus type 1 (HIV-1) is activated by superinfection with CMV or herpes virus [23]. It was recently revealed that long-term oxidative stress on human lymphoid cell lines infected with HIV-1 in vitro leads to upregulation of the HIV-1 promoter gene [24]. Therefore, induction of oxygen radical production in virus-infected hosts may be attributable not only to tissue damage caused directly as well as indirectly by the virus but also to enhancement of viral replication or reactivation in some viral diseases.
GENERATION OF NO IN VIRUS INFECTION
Induction of iNOS in viral infections is now known for a wide range of viruses with different organ tropisms, including neuro-, pneumo-, and cardio-tropic viruses such as Borna disease virus, herpes simplex virus type 1 (HSV-1), rabies virus, influenza virus, Sendai virus, and coxsackievirus [18, 20, 25-30]. It is potentially of interest that iNOS was expressed in the brain tissue of a patient with HIV-1 encephalitis [31].
We then realized that the above story of superoxide toxicity is not so simple, as shown in Fig. 5 (a and b), when both XO and NOS are induced simultaneously in the influenza virus infected mouse lung [27]. The iNOS expression was identified mainly localized in macrophages and bronchial epithelial cells in the lung [18, 20, 27].
The induction of iNOS is shown to involve the action of IFN-gamma [27] and TNF-alpha [20]. Figure 5 (c and d) shows that anti-IFN-gamma antibodies, if added to the BALF from the infected mice, suppressed NOS induction caused by the BALF in RAW264 cells in vitro. Influenza viral particles per se possess the capacity to induce iNOS in the macrophage, although it needs identification of actual chemical entity of the viral components. This phenomenon of iNOS induction by virus is similar to LPS or teichoic acid of bacteria in the upregulation of iNOS induction [2, 3].Fig. 5. Induction of iNOS in mouse lung after influenza virus infection (2 × LD50 of influenza virus A/Kumamoto/Y5/67(H2N2)). a) NOS activity induced in the virus infected lungs. b) Expression of iNOS mRNA. mRNA expressions of iNOS and G3PDH were analyzed by RT-PCR with Southern blotting. c) Induction of IFN-gamma (IFN-gamma in plasma (1) and s-BALF (2)) in influenza virus infection in mice. d) NOS induction potential of the supernatant of BALF (s-BALF) in RAW264 cells in culture. NOS induction in RAW264 cells in culture was assessed by measuring nitrite released in the culture after stimulation with serially diluted s-BALF and virus: 1) control; 2) normal s-BALF; 3) virus-infected s-BALF at dilution 10, 50, and 100; 4) virus-infected s-BALF at dilution 10 and 50 + anti-IFN-gamma antibodies; 5) influenza virus at different concentrations (log(PFU) values are 6, 7, and 8). Data are means ± SEM (n = 4) [27].
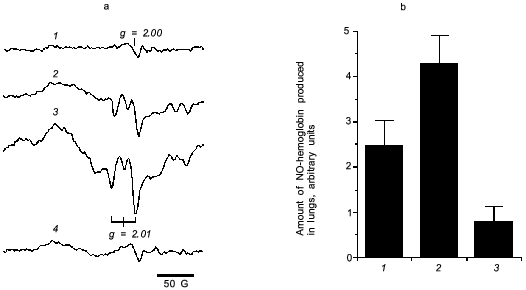
For direct examination of NO production in the virus infected mouse lung, we measured NO-hemoglobin signal using electron paramagnetic resonance (EPR) spectroscopy, as shown in Fig. 6.
It is known that a rapid coupling reaction of NO and O2·- forms uniquely reactive peroxynitrite (ONOO-):Fig. 6. a) EPR spectra of NO-hemoglobin from lungs of control (uninfected) mice (1) and mice infected with influenza virus (2-4) without (2) or with treatment by polyvinylalcohol (PVA)--SOD (3) and L-NMMA (4). b) The amount of NO-hemoglobin was quantified by double integration of each spectrum and corrected with hemoglobin content: 1) virus infection alone; 2) infection + PVA--SOD; 3) infection + L-NMMA. Data are means ± SD (n = 5) [27].
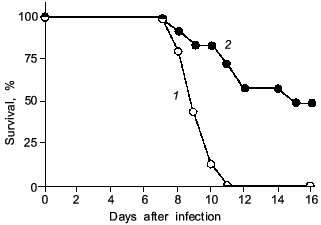
It is noteworthy that a significant improvement in survival rate was obtained with L-NMMA treatment of the influenza-virus infected animals [27] (Fig. 7). Similar results were obtained by Kreil and Eibl regarding the effect of NOS inhibition on tick-borne encephalitis (TBE)-virus infection in mice [29]. In their report, excessive NO generation in murine macrophages did not result in inhibition of TBE-virus replication in vitro. Also, treatment of the TBE-virus infected mice with the NOS inhibitor aminoguanidine produced a significantly increased survival time.
We recently examined the effect of NOS inhibition with L-NMMA on HSV-1-induced encephalitis in rats. Although an antiproliferative action of NO against HSV was described for cells in culture [32, 33], our results in vivo indicate that L-NMMA suppression of excessive production of NO in the central nervous system (CNS) of HSV-1-infected animals resulted in improvement in neuronal damage, but suppression of NO generation did not affect virus propagation in the CNS (our unpublished observation).Fig. 7. The effect of L-NMMA treatment (2) on survival rate of mice after influenza virus infection ((1) is control). The 2×LD50 dose of the virus was given as described in Fig. 1 legend. The mice were treated intraperitoneally with L-NMMA (2 mg/mouse in 0.2 ml PBS) every 24 h from day 3 to 7 (n = 20) [27].
An important observation was reported recently by Adler et al. regarding the effect of NO in HSV-1-induced pneumonia [30], in which they investigated the effect of iNOS inhibition by L-NMMA treatment of HSV-1-infected mice on various pathological parameters of HSV-1-induced pneumonia such as histopathological changes of the lung, the pulmonary function (compliance of the lung), the mortality due to pneumonia, and the viral yield in the lung. They showed that L-NMMA treatment led to a significant improvement of pneumonia despite increased virus growth in the lung. Thus it was concluded that the viral pathogenesis of HSV-1-induced pneumonia is closely related to the NO-mediated inflammatory response of the host rather than direct effect of HSV-1 replication in the lung. This notion is essentially consistent with our interpretation of the role of NO in the pathogenesis of influenza virus-induced pneumonia in mice.
These events are similarly demonstrated in pulmonary granulomatous inflammation in rats after intratracheal instillation of silica particles and zymosan obtained from yeast [34]. By the same token, chronic infections with H. pylori in the human stomach [9] and with some parasite (O. viverrini) [14] may bring about excessive NO production and thus lead to carcinogenesis. In this context, the granuloma formation and cancer formation have a common pathogenic event in view of NO generation.
BIOLOGICAL EFFECTS OF PEROXYNITRITE
Based on the above-mentioned results, overproduction of NO in virus infection and chronic inflammation may contribute to pathological consequences if it is accompanied by the production of superoxide. This notion is supported by the formation of peroxynitrite and its unique biochemical properties.
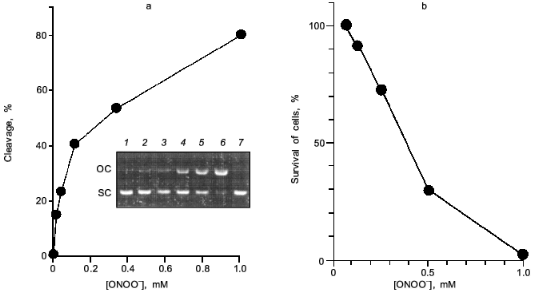
Peroxynitrite is much more reactive than NO or superoxide [35-38]. It will cause diverse chemical reactions in biological system including nitration of tyrosine residues of proteins [39, 40], triggering of lipid peroxidation [41, 42], inactivation of aconitases [43, 44], inhibition of mitochondrial electron transport [45], and oxidation of biological thiol compounds [46]. Peroxynitrite also exerts a potent DNA cleaving activity [10, 11] (Fig. 8a). These chemical reactions of peroxynitrite will lead to a profound biological consequence such as apoptosis, and even mutation of various cells [38, 47-52]. The direct cytotoxic action of peroxynitrite against cells in culture is shown in Fig. 8b. The nitration of tyrosine residues in cells was recently suggested to compromise phosphorylation or adenylation of proteins, resulting in impairment of intracellular signal transduction [53, 54]. The biological relevance of peroxynitrite should be further emphasized by the recent finding that chemical reactivity of peroxynitrite are modulated or potentiated by carbon dioxide (CO2) or carbonate ion [55], which exists in physiological fluids at about 1.2 mM [56].
In this regards, Xia and Zweier reported the intriguing finding that effective peroxynitrite production was observed in iNOS-expressed murine macrophages when they were cultured under L-arginine-depleted conditions. However, peroxynitrite formation was not observed appreciably in L-arginine-supplemented culture [57]. This suggests that the insufficient substrate (L-arginine) supply for NOS leads to excessive and effective production of peroxynitrite rather than NO per se from iNOS.Fig. 8. Biological effect of peroxynitrite. a) DNA cleavage. pUC19 plasmid DNA was incubated with various concentrations of peroxynitrite for 30 min at 37°C. DNA cleavage was estimated by measuring morphological transition of plasmid DNA from supercoiled form (SC) to open circular form (OC). Lanes: 1) DNA (0.1 mg) alone; 2-6) DNA treated with ONOO- at 13, 38, 110, 330, and 1.0 mM, respectively; 7) DNA with decomposed ONOO- (1.0 mM). b) Cytotoxic effect on a rat glial cell line (C6 cells). Assay was based on MTT methods in a multiplate Petri dish.
Moreover, we recently found that peroxynitrite activated human neutrophil procollagenase (matrix metalloproteinase 8, MMP-8), which is critically involved in tissue disintegration and remodeling under physiological as well as pathological conditions such as inflammation and infection [58, 59]. In addition to activation of MMP-8, peroxynitrite readily inactivates both tissue inhibitor for MMP (TIMMP) and alpha1-proteinase inhibitor, a major proteinase (neutrophil elastase) inhibitor in human plasma [60-62]. Thus, peroxynitrite seems to accelerate tissue degradation and contribute to the pathogenesis of various diseases. It is also reported that peroxynitrite activates cyclooxygenase, which is a key enzyme in the production of the potent inflammatory mediators, the prostaglandins [63].
BACTERIAL INFECTION AND GENERATION OF FREE RADICALS
Bacterial infection per se results in induction of both XO and iNOS in parallel with cytokine induction similar to virus infection as a part of the host defense response. This is a similar phenomenon to the induction of iNOS by injecting LPS [1, 3], because LPS is a component of the outer membrane of gram-negative bacteria.
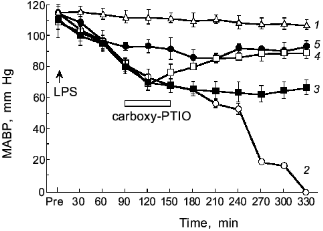
The intravenous injection of LPS results in hypotensive shock caused by NO generation as shown in Fig. 9 [3]. If we scavenge NO, the shock of the LPS-treated rats will be blocked as evidenced by the therapeutic effect of NO scavenger, PTIO (2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide): the higher the dose of PTIO, the better the antihypotensive effect [3, 64]. Thus, the protective effect of NO scavenger PTIO against the hypotension caused by endotoxin was demonstrated clearly.
Salmonella typhimurium infection in mice showed induction of XO and iNOS [65, 66]. In addition, peroxynitrite formation was observed around the granulomatous lesions in the Salmonella-infected liver, based on the positive immunostaining of the tissues with antinitrotyrosine antibody similar to the influenza model. The increased cancer incidence in Salmonella-infected carrier in humans has been reported by an earlier epidemiological study [13]. Therefore, the free radical and peroxynitrite generation in the S. typhimurium infection in mice may suggest another example of a link between chronic bacterial infection and cancer.Fig. 9. Effects of carboxy-PTIO on the mean arterial blood pressure (MABP) of rats with endotoxin shock. Various concentrations of carboxy-PTIO (0.056 (3), 0.17 (4), and 1.7 mg/kg per min (5)) were administered by continuous intravenous infusion for 1 h at the rate of 6 ml/h, 90 min after intravenous injection of E. coli LPS (10 mg/kg); 1) vehicle-treated control; 2) LPS-treated control. Data are means ± SEM (n = 5) [3].
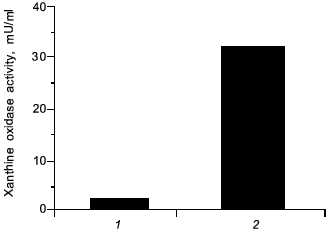
In view of carcinogenesis and DNA damage, we have found a similar effect by administration of a chemical carcinogen (carbon tetrachloride) per se in rats which induced elevated the level of XO by 25-fold higher than non-treated control, similar to the microbial infection (Fig. 10), which then will generate superoxide. It is possible that NO might similarly be formed and damaged the tissues, and carcinogenesis will be caused by carbon tetrachloride accordingly.
Fig. 10. Induction of xanthine oxidase (XO) by carbon tetrachloride. Increased activity of XO in serum from rats with hepatitis (2) was induced 24 h after oral administration of carbon tetrachloride ((1) is control, healthy rats).
ENHANCEMENT OF TUMOR GROWTH BY NO
One more issue relating to NO in cancer is its involvement in tumor growth. We and others have previously demonstrated that NO functions as a vascular permeability factor [67-69], and most tumor tissue exhibits enhanced vascular permeability. This means tumor blood vessels are very leaky, and even albumin and other macromolecules are able to traverse out of the blood vessels. Other important factors that affect the vascular permeability of solid tumor are bradykinin [5, 70, 71] and a vascular permeability factor (VPF; also called as vascular endothelial growth factor, VEGF) [72].
Interestingly, Ziche et al. recently reported that VEGF induces angiogenesis via formation of NO [73]. Angiogenesis leading to hypervasculature is often observed during rapid tumor growth. Consequently, it is plausible that the angiogenic potential as well as the vascular permeability enhancing effect of NO may facilitate rapid growth of solid tumors, which have great demands for various nutrient factors. A similar beneficial effect of NO on tumor growth was clearly demonstrated recently in tumor-bearing mice in which human adenocarcinoma cells, which overexpressed iNOS, were implanted [74].
We now know that the inhibition or scavenging of NO or bradykinin results in suppression of permeability and tumor growth, whereas vascular permeability is proportional to the tumor growth [68, 69, 75, 76]. Specifically, we examined the effect of extravasation by injecting inhibitors of NO synthase and NO scavenger. For instance, L-NAME (Nomega-nitro-L-arginine methyl ester) as well as PTIO can effectively inhibit both the extravasation and tumor growth [68, 75, 77].
Furthermore, bradykinin is known to activate a constitutive isoform of NOS (endothelial NOS) [4]. Because bradykinin is generated effectively in tumors [70, 71], bradykinin-NO interplay may become an important issue in tumor growth.
GENERAL CONSIDERATION AND CONCLUDING REMARKS
NO is oxidized by one way or another, and then various nitrogen oxide derivatives will be generated. Incidentally, it has been known as early as 1960s that nitrous acid damages nucleic acids by deamination or nitration [78]; therefore, it is natural that a central theme of carcinogenesis is free radicals and nitrogen oxides. In particular, hydroxyl radical as well as peroxynitrite formed by the reactions of NO and superoxide will be the most crucial molecular species.
Classic text books in oncology or pathology describe three completely different causes of cancer. They are chemical carcinogens (or chemicals), tumor viruses, and exposure to radiation such as X-ray, gamma-ray, and ultraviolet light (cf. Fig. 1). The last one, radiation, is the oldest known factor for generating free radicals in radiation physics. Hydroxyl radical, superoxide or singlet oxygen are generated by the electromagnetic interaction between water or oxygen molecules, which reacts with nucleic acids, proteins and other vital molecules. Damage to nucleic acids is known as a cause of mutation and consequently cancer. Now, such radicals as superoxide became well known to exist in biological systems. We now have ample evidence that microbial or parasitic infection can generate free radicals as well as reactive oxygen and nitrogen oxide intermediate as in inflammatory conditions as described in this article. In the different settings, we have observed chemical carcinogens, e.g., carbon tetrachloride as mentioned above and heterocyclic amines can generate superoxide in the presence of cytochrome P-450 system or cytochrome P-450 reductase [79]. Thus, one of the most well studied carcinogen (heterocyclic amine) is a source of superoxide generation, and incidentally, 8OHG generation (as a result of reaction with hydroxyl radical) became well established [80]. In this context, preventive measures for cancer can be recommended to minimizing excessive exposure to free radicals in vivo either by dietary considerations, e.g., diet high in flavonoids, or by adequate antiinflammatory regimens, or control of chronic infections.
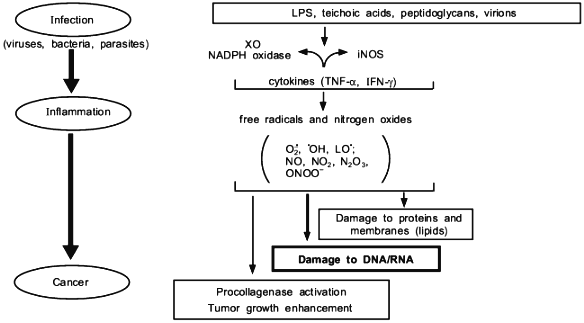
We have presented here examples of free radical generation in viral and bacterial infection which would result in similar consequence to that of radiation and chemical carcinogens. The enzymes induced to generate free radicals upon infection so far demonstrated include XO and NADPH oxidase for superoxide, and NOS for NO. An extremely rapid reaction between O2·- + NO yields ONOO- which is more reactive and DNA damaging than O2·- or NO alone. In any event, the free radical hypothesis in carcionogenesis seems to unify all different carcinogenic mechanisms as summarized in Fig. 11.
We thank Ms. R. Yoshimoto for preparing and editing the manuscript. This work is supported by a Grant-in-Aid from the Ministry of Education, Science, Sports and Culture of Japan, and a grant from the Ministry of Health and Welfare of Japan for surveys and research on specific diseases.Fig. 11. Various events involving free radicals in infection and inflammation that lead to cancer.
REFERENCES
1.Nathan, C. (1992) FASEB J., 6,
3051-3064.
2.De Kimpe, S. J., Kengatharan, M., Thiemermann, C.,
and Vane, J. R. (1995) Proc. Natl. Acad. Sci. USA, 92,
10359-10363.
3.Yoshida, M., Akaike, T., Wada, Y., Sato, K., Ikeda,
K., Ueda, S., and Maeda, H. (1994) Biochem. Biophys. Res.
Commun., 202, 923-930.
4.Palmer, R. M. J., Ashton, D. S., and Moncada, S.
(1988) Nature, 333, 664-666.
5.Maeda, H., Akaike, T., Wu, J., Noguchi, Y., and
Sakata, Y. (1996) Immunopharmacol., 33, 222-230.
6.Harris, C. C. (1991) Cancer Res. (Suppl.),
51, 5023S-5044S.
7.Haugen, H., and Harris, C. C. (1990) in Handbook
of Experimental Pharmacology (Cooper, C. S., and Grover, P. L.,
eds.) Vol. 94, Springer-Verlag, Berlin, pp. 249-268.
8.Yuspa, S. H., and Poivier, M. C. (1988) Adv.
Cancer Res., 50, 25-70.
9.Pignatelli, B., Bancel, B., Esteve, J., Malaveille,
C., Calmels, S., Correa, P., Patricot, L. M., Laval, M., Lyandrat, N.,
Smit, F., and Ohshima, H. (1996) in Recent Advances in
Gastroenterological Carcinogenesis (Tahara, E., Sugimachi, K., and
Oohara, T., eds.) Monduzzi Editore S.p.a, Bologna, pp. 1221-1225.
10.Inoue, S., and Kawanishi, S. (1995) FEBS
Lett., 311, 86-88.
11.Yermilov, V., Yoshie, Y., Rubio, J., and Ohshima,
H. (1996) FEBS Lett., 399, 67-70.
12.Baik, S.-C., Youn, H.-S., Chung, M.-H., Lee,
W.-K., Cho, M.-J., Ko, G.-H., Park, C.-K., Kasai, H., and Rhee, K.-H.
(1996) Cancer Res., 56, 1279-1282.
13.Caygill, C. P. J., Hill, M. J., Braddick, M., and
Sharp, J. C. M. (1994) Lancet, 343, 83-84.
14.Ohshima, H., Bandaletova, T. Y., Brouet, I.,
Bartsch, H., Kirby, G., Ogunbiyi, F., Vantanasapt, V., and Pipitgool,
V. (1994) Carcinogenesis, 15, 271-275.
15.Uemura, Y., Tokunaga, M., Arikawa, J., Yamamoto,
N., Hamasaki, Y., Tanaka, S., Sato, E., and Land, C. E. (1994)
Cancer Epidemiol. Biomarkers and Prevention, 3,
607-611.
16.Oda, T., Akaike, T., Hamamoto, T., Suzuki, F.,
Hirano, T., and Maeda, H. (1989) Science, 244,
974-976.
17.Akaike, T., Ando, M., Oda, T., Doi, T., Ijiri,
S., Araki, S., and Maeda, H. (1990) J. Clin. Invest., 85,
739-745.
18.Akaike, T., Suga, M., and Maeda, H. (1998)
Proc. Soc. Exp. Biol. Med., 217, 64-73.
19.Maeda, H., and Akaike, T. (1991) Proc. Soc.
Exp. Biol. Med., 198, 721-727.
20.Akaike, T., and Maeda, H. (1998) in Nitric
Oxide in Infection (Fang, F. C., ed.) Plenum Publishing
Corporation, New York, London, Moscow, in press.
21.Ikeda, T., Shimokata, K., Daikoku, T., Fukatsu,
T., Tsutsui, Y., and Nishiyama, Y. (1993) Arch. Virol.,
127, 11-24.
22.Sidwell, R. W., Huffman, J. H., Bailey, K. W.,
Wong, M. H., Nimrod, A., and Panet, A. (1996) Antimicrob. Agents
Chemother., 40, 2626-2631.
23.Mosca, J. D., Bednarik, D. P., Raj, N. B. K.,
Rosen, C. A., Sodroski, J. G., Haseltine, W. A., and Pitha, P. M.
(1987) Nature, 325, 67-70.
24.Kurata, S.-I. (1996) J. Biol. Chem.,
271, 21798-21802.
25.Koprowski, H., Zheng, Y. M., Heber-Katz, E.,
Fraser, N., Rorke, L., Fu, Z. F., Hanlon, C., and Dietzschold, B.
(1993) Proc. Natl. Acad. Sci. USA, 90, 3024-3027.
26.Akaike, T., Weihe, E., Schaefer, M., Fu, Z. F.,
Zheng, Y. M., Vogel, W., Schmidt, H., Koprowski, H., and Dietzschold,
B. (1995) J. NeuroVirol., 1, 118-125.
27.Akaike, T., Noguchi, Y., Ijiri, S., Setoguchi,
K., Suga, M., Zheng, Y. M., Dietzschold, B., and Maeda, H. (1996)
Proc. Natl. Acad. Sci. USA, 93, 2448-2453.
28.Mikami, S., Kawashima, S., Kanazawa, K., Hirata,
K., Katayama, Y., Hotta, H., Hayashi, Y., Ito, H., and Yokoyama, M.
(1996) Biochem. Biophys. Res. Commun., 220, 983-989.
29.Kreil, T. R., and Eibl, M. M. (1996)
Virology, 219, 304-306.
30.Adler, H., Beland, J. L., Del-Pan, N. C., Kobzik,
L., Brewer, J. P., Martin, T. R., and Rimm, I. J. (1997) J. Exp.
Med., 185, 1533-1540.
31.Bukrinsky, M. I., Nottet, H. S. L. M.,
Schmidtmayerova, H., Dubrovsky, L., Flanagan, C. R., Mullins, M. E.,
Lipton, S. A., and Gendelman, H. E. (1995) J. Exp. Med.,
181, 735-745.
32.Karupiah, G., Xie, Q., Buller, R. M. L., Nathan,
C., Duarte, C., and MacMicking, J. D. (1993) Science,
261, 1445-1448.
33.Croen, K. D. (1993) J. Clin. Invest.,
91, 2446-2452.
34.Setoguchi, K., Takeya, M., Akaike, T., Suga, M.,
Hattori, R., Maeda, H., Ando, M., and Takahashi, K. (1996) Am. J.
Pathol., 149, 2005-2022.
35.Beckman, J. S., Beckman, T. W., Chen, J.,
Marshall, P. A., and Freeman, B. A. (1990) Proc. Natl. Acad. Sci.
USA, 87, 1620-1624.
36.Pryor, W. A., and Squadrito, G. L. (1995) Am.
J. Physiol., 268, L699-L722.
37.Beckman, J. S., and Koppenol, W. H. (1996) Am.
J. Physiol., 271, C1424-C1437.
38.Rubbo, H., Darley-Usmar, V., and Freeman, B. A.
(1996) Chem. Res. Toxicol., 9, 809-820.
39.Beckman, J. S., Ye, Y. Z., Anderson, P. G., Chen,
J., Accavitti, M. A., Tarpey, M. M., and White, C. R. (1994) Biol.
Chem. Hoppe Seyler, 375, 81-88.
40.Haddad, I. Y., Pataki, G., Hu, P., Galliani, C.,
Beckman, J. S., and Matalon, S. (1994) J. Clin. Invest.,
94, 2407-2413.
41.Radi, R., Beckman, J. S., Bush, K. M., and
Freeman, B. A. (1991) Arch. Biochem. Biophys., 288,
481-487.
42.Haddad, I. Y., Ischiropoulos, B., Holm, B. A.,
Beckman, J. S., Baker, J. R., and Matalon, S. (1993) Am. J.
Physiol., 265, L555-L564.
43.Castro, L., Rodriguez, M., and Radi, R. (1994)
J. Biol. Chem., 269, 29409-29415.
44.Hausladen, A., and Fridovich, I. (1994) J.
Biol. Chem., 269, 29405-29408.
45.Radi, R., Rodriguez, M., Castro, L., and Telleri,
R. (1994) Arch. Biochem. Biophys., 308, 89-95.
46.Radi, R., Beckman, J. S., Bush, K. M., and
Freeman, B. A. (1991) J. Biol. Chem., 266, 4244-4250.
47.Dawson, V. L., Dawson, T. M., Uhl, G. R., and
Snyder, S. H. (1993) Proc. Natl. Acad. Sci. USA, 90,
3256-3259.
48.Zhu, L., Gunn, C., and Beckman, J. S. (1992)
Arch. Biochem. Biophys., 298, 452-457.
49.Bonfoco, E., Krainc, D., Ankarcrona, M.,
Nicotera, P., and Lipton, S. A. (1995) Proc. Natl. Acad. Sci.
USA, 92, 7162-7166.
50.Troy, C. M., Derossi, D., Prochiantz, A., Greene,
L. A., and Shelanski, M. L. (1996) J. Neurosci., 16,
253-261.
51.Estevez, A. G., Radi, R., Barbeito, L., Shin, J.
T., Thompson, J. A., and Beckman, J. S. (1995) J. Neurochem.,
65, 1543-1550.
52.Ischiropoulos, H., Al-Mehdi, A., and Fisher, A.
B. (1995) Am. J. Physiol., 269, L158-L164.
53.Berlett, B. S., Friguet, B., Yim, M. B., Chock,
P. B., and Stadtman, E. R. (1996) Proc. Natl. Acad. Sci. USA,
94, 1776-1780.
54.Kong, S. K., Yim, M. B., Stadtman, E. R., and
Chock, P. B. (1996) Proc. Natl. Acad. Sci. USA, 93,
3377-3382.
55.Uppu, R. M., Squadrito, G. L., and Pryor, W.
(1996) Arch. Biochem. Biophys., 327, 335-343.
56.Garrett, R. H., and Grisham, C. M. (1995) in
Biochemistry, Saunders College Publishing, Fort Worth, pp.
32-54.
57.Xia, Y., and Zweier, J. L. (1997) Proc. Natl.
Acad. Sci. USA, 94, 6954-6958.
58.Okamoto, T., Akaike, T., Nagano, T., Miyajima,
S., Suga, M., Ando, M., Ichimori, K., and Maeda, H. (1997) Arch.
Biochem. Biophys., 342, 261-274.
59.Okamoto, T., Akaike, T., Suga, M., Tanase, S.,
Horie, H., Miyajima, S., Ando, M., Ichinose, Y., and Maeda, H. (1997)
J. Biol. Chem., 272, 6059-6066.
60.Moreno, J. J., and Pryor, W. (1992) Chem. Res.
Toxicol., 5, 425-431.
61.Frears, E. R., Zhang, Z., Blake, D. R.,
O'Connell, J. P., and Winyard, P. G. (1996) FEBS Lett.,
381, 21-24.
62.Whiteman, M., Tritschler, H., and Halliwell, B.
(1996) FEBS Lett., 379, 74-76.
63.Landino, L. M., Crews, B. C., Timmons, M. D.,
Morrow, J. D., and Marnett, L. J. (1996) Proc. Natl. Acad. Sci.
USA, 93, 15069-15074.
64.Akaike, T., Yoshida, M., Miyamoto, Y., Sato, K.,
Kohno, M., Sasamoto, K., Miyazaki, K., Ueda, S., and Maeda, H. (1993)
Biochemistry, 32, 827-832.
65.Umezawa, K., Ohnishi, N., Tanaka, K., Kamiya, S.,
Koga, Y., Nakazawa, H., and Ozawa, A. (1995) Infect. Immun.,
63, 4402-4408.
66.Umezawa, K., Akaike, T., Fujii, S., Suga, M.,
Setoguchi, K., Ozawa, A., and Maeda, H. (1997) Infect. Immun.,
65, 2932-2940.
67.Maeda, H., Noguchi, Y., Sato, K., and Akaike, T.
(1994) Jpn. J. Cancer Res., 85, 331-334.
68.Doi, K., Akaike, T., Horie, H., Noguchi, Y.,
Fujii, S., Beppu, T., Ogawa, M., and Maeda, H. (1996) Cancer,
77, 1598-1604.
69.Jenkins, D. C., Charles, I. G., Thomsen, L. L.,
Moss, D. W., Holmens, L. S., Baylis, S. A., Rhodes, P., Westmore, K.,
Emson, P. C., and Moncada, S. (1995) Proc. Natl. Acad. Sci. USA,
92, 4392-4396.
70.Maeda, H., Matsumura, Y., and Kato, H. (1988)
J. Biol. Chem., 263, 16051-16054.
71.Matsumura, Y., Kimura, M., Yamamoto, T., and
Maeda, H. (1988) Jpn. J. Cancer Res., 79, 1327-1334.
72.Senger, D. R., Galli, S. J., Dvorak, A. M.,
Perruzzi, C. A., Harvey, V.-S., and Dvorak, H. F. (1983)
Science, 219, 983-985.
73.Ziche, M., Morbidelli, L., Choudhuri, R., Zhang,
H.-T., Donnini, S., Granger, H. J., and Bicknell, R. (1997) J. Clin.
Invest., 99, 2625-2634.
74.Jenkins, D. C., Charles, I. G., Thomsen, L. L.,
Moss, D. W., Holmes, L. S., Baylis, S. A., Rhodes, P., Westmore, K.,
Emson, P. C., and Moncada, S. (1995) Proc. Natl. Acad. Sci. USA,
92, 4392-4396.
75.Wu, J., Akaike, T., and Maeda, H. (1998)
Cancer Res., in press.
76.Kallinowski, F., Schlenger, K. H., Runkeels, S.,
Kloes, M., Stohner, M., Okunieff, P., and Vaupel, P. (1989) Cancer
Res., 49, 3759-3764.
77.Doi, K., Akaike, T., Fujii, S., Ikebe, N., Beppu,
T., Ogawa, M., and Maeda, H. (1998) in The Biology of Nitric
Oxide (Moncada, S., Toda, N., Maeda, H., and Higgs, E. A., eds.)
Pt. 6, Portland Press, London, in press.
78.Fraenkel-Conrat, H. (1964) Sci. Am.,
211, 47-54.
79.Sato, K., Akaike, T., Kojima, Y., Ando, M.,
Nagao, M., and Maeda, H. (1992) Jpn. J. Cancer Res., 83,
1204-1209.
80.Kamiya, H., Murata-Kamiyu, N., Koizumi, S.,
Inoue, H., and Nishimura, E. (1995) Carcinogenesis, 16,
883-889.