REVIEW: NO-Synthase and Nitrite-Reductase Components of Nitric Oxide Cycle
V. P. Reutov* and E. G. Sorokina
Institute of Higher Nervous Activity and Neurophysiology, Russian Academy of Sciences, ul. Butlerova 5a, Moscow, 117865 Russia; fax: (095) 338-8500* To whom correspondence should be addressed.
Received July 17, 1997
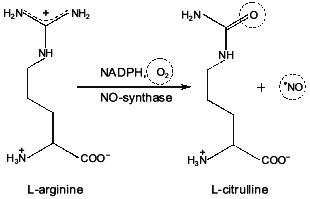
On the basis of our own experimental data and analysis of data from the literature the existence of nitric oxide cycle in mammals is substantiated. Two components underlie the nitric oxide cycle: 1) the reaction catalyzed by NO-synthases (constitutive, inducible, and endothelial--NOS-I, -II, and -III); and 2) the nitrite-reductase reactions catalyzed by electron-donor systems with the participation of NADH, NADPH, flavoproteins, and heme-containing proteins. In mammalian cells NO is enzymatically formed from terminal guanidine nitrogen of L-arginine by a family of at least three distinct NOS isoenzymes. As a result of nonenzymatic/enzymatic NO oxidation, NO2- and NO3- ions are formed: L-Arg --> NO --> NO2-/NO3-. The reduction of NO2- ions to NO occurs via the nitrite-reductasereaction: NO2- + e- --> NO. The reduction of NO2- ions to NO is realized by electron-donor systems with the participation of NADH, NADPH, flavoproteins, and cytochrome oxidase in mitochondria and by NADH, NADPH, flavoproteins, and cytochrome P-450 in endoplasmic reticulum. In erythrocytes the reduction of NO2- ions to NO is catalyzed by electron-donor systems with participation of NADH, NADPH, flavoproteins, and deoxy-hemoglobin. The role of ascorbic acid and reduced glutathione should be noted among low-molecular-weight compounds. Thus, the presence of the nitric oxide cycle provides the cyclic transformation as follows: L-arginine --> NO --> NO2-/NO3- --> NO.
KEY WORDS: nitric oxide cycle, Hb--NO and Mb--NO-complexes, NO2- and NO3- ions, hypoxia
Abbreviations: IP3) inositol trisphosphate; cADPR) cyclic adenosine diphosphate-ribose; DAG) diacylglycerol; AA) arachidonic acid; EDRF) endothelium-derived relaxation factor; BH4) tetrahydrobiopterin.
Indeed, the history of oxygen is the history of life!
L. Traube
... Any biological study is justified only when it has a near or
distant, but without fail, evolutionary exit.
N. V. Timofeyev-Resovsky
At present the problem of NO and nitrocompounds attracts attention of
different kinds of biologists and medical specialists [1-9]. Analysis of the data from
the literature of the 1980-90s suggests that studies on
medical-biological aspects of NO embrace an unprecedentedly wide region
at the boundary between biochemistry and molecular biology, physiology
and cellular biology, pharmacology and toxicology [10-20]. Why did such a simple,
at the first sight, molecule get into the focus of attention of
researchers?
Within the last decade NO has become known to participate in regulation of intracellular concentrations of Ca2+ in mammals [21-23]. One of the mechanisms of regulation is related to NO synthesis activation, an increase in intracellular concentration of cGMP, and the activation of Ca2+-pumps of the endoplasmic reticulum through a system of G-kinases [21, 22]. Another mechanism may be regulation of release of Ca2+ from its pool that is insensitive to inositol trisphosphate (IP3) but sensitive to Ca2+ and cyclic adenosine diphosphate-ribose (cADPR) [24, 25]. Activation of ADP-ribosyl-transferase with the participation of NO [26] can influence the mechanism of release of Ca2+ from the second pool, which is insensitive to IP3. And finally, to some researchers’ minds [27], the formation of NO+, being one of the intermediates of NO metabolism products, can affect the permeability of Ca2+ channels. The discovery of these properties in NO made it possible to consider this compound to be among such secondary messengers as cAMP, cGMP, Ca2+, IP3, diacylglycerol (DAG), cADPR, and arachidonic acid (AA) [28-36].
In this paper we have analyzed the data from the literature and the results of twenty years of our own research with the goal of elucidating the role of NO in biological systems. Our results and analysis of the literature [37-46] suggest that in blood and cells of different tissues, NO participates in a metabolic transformation in the course of which nitro- and nitroso-compounds able to be transformed back into NO are formed. This permitted us to be the first to conclude that NO-synthase and nitrite-reductase components in mammals form the nitric oxide cycle [47, 48]. But although the NO-synthase component is intensively studied [49-58], the nitrite-reductase component still remains in the shade, though its presence is revealed by researchers each time the question of the nature of endothelium-derived relaxation factor (EDRF) of blood vessels [59-63] and regulation mechanisms of NO content in the body under physiological conditions and in hypoxias of different genesis is under consideration [47, 48, 64, 65]. So, the main attention in our paper is given to consideration of the less known mechanisms of restoration of NO2- to NO and the nitrite-reductase capability of heme-containing proteins.
NO-SYNTHASE COMPONENT OF THE NITRIC OXIDE CYCLE: NO SYNTHESIS IN
THE PRESENCE OF OXYGEN
Nitric oxide synthases are now known not to be a single enzyme, but a family or a group of enzymes which are able to form NO from L-arginine. When classifying nitric oxide synthases, three types or three isoenzymes coded by different genes are found [66-69].
Type I NO-synthase is present in neurons in the brain and is often called neuronal constitutive NO synthase [70-73]. The activity of constitutive NO-synthase is the highest in the neurons of the cerebellum and in astroglia. This enzyme is a homodimer (consisting of two equal subunits) with the molecular subunit mass of 160 kD and is characterized by reversible binding with calmodulin. There is evidence on that over 50% of the amino acid sequence of neuronal constitutive NO-synthase are identical to cytochrome P-450 reductase [69, 73]. This enzyme is regulated by Ca2+. The Michaelis constant is 2-7 µM. The posttranslation modification of this enzyme is known to be capable of considerable modulating of its activity as a result of phosphorylation.
Type II NO-synthase was first isolated from macrophages. It is a homodimer with molecular mass of 130 kD [74-76]. The Michaelis constant is 3-32 µM, and the maximum rate of L-arginine oxidation and NO formation can reach 1600 nmoles/min per mg. This enzyme is mainly in the soluble form and is an inducible isoform of NO-synthase (iNOS). Rather recently iNOS has been purified [74, 75] and cloned [77, 78] from interferon-gamma and lipopolysaccharide-activated mouse macrophages.
The primary structure obtained by analyzing cDNA differs from the constitutive NO-synthase cloned from cerebellum cells and endothelial cells. In contrast to the constitutive enzymes, this enzyme is less dependent on Ca2+ or calmodulin. However, inducible NO-synthase, like the constitutive one, contains a calmodulin-binding region. There are some data indicating the relative calmodulin-independence of this enzyme to be due to the fact that calmodulin is firmly bound to this NO-synthase isoform. In recent years iNOS has been found to be present not only in macrophages, but also in some glial cells, for example, in brain microglia [69, 79].
Type III NO-synthase is typical for endothelial cells. The molecular mass of this enzyme is 133 kD [69, 79, 80]. Like type I NO-synthase, this enzyme is characterized by reversible binding with calmodulin and its activity depends on the intracellular concentration of Ca2+. The main differences of type III NO-synthase from type I are at the N-terminal of the protein molecule. Nevertheless, 57% of the amino acid sequence of type III NO-synthase are identical to type I NO-synthase. This enzymatic isoform has also been found in the neurons in the brain (hippocampus). The Michaelis constant is 3 µM and Vmax is 15 nmoles/min per mg. The enzyme activity is modulated by posttranslational modification (phosphorylation and myristoylation). This enzyme can be in both soluble and membrane-bound forms. Localization of this enzyme in the plasmatic membrane appears to be of substantial significance for the signal transfer mechanism (transduction) with participation of NO during a shift in tension arising upon acceleration of the blood flow. How is NO formed in the course of the NO-synthase reaction?
MECHANISM OF NO FORMATION WITH THE PARTICIPATION OF
NO-SYNTHASES
A hypothetical model for explaining the structure and functions of NO-synthase has been proposed [69]. According to this model, the NO-synthase dimer formation occurs by an intermolecular contact in the calmodulin-binding domain (Fig. 1). As a result of the contact, a dimeric structure is formed where the head of one monomer is associated with the tail of the other monomer. Arginine and tetrahydrobiopterin (BH4) are assumed to be associated in the region near the NH2 terminal. The authors believe that BH4 can either enter this process immediately, or it is involved in allosteric regulation of NO-synthase. It has been demonstrated that 1.5 mole NADPH is used for synthesis of one molecule of NO. However, the role of BH4 is still to be explained. Flavin cofactors FAD and FMN as well as the cytochrome P-450 domain of NO-synthase act as electron carriers from NADPH to molecular oxygen (oxidase domain). It is presumed that the hydroxylated form of L-arginine with the participation of O2- and the cytochrome P-450 domain heme is converted to L-citrulline with simultaneous formation of NO [69].
It should be noted in particular that molecular oxygen is used for NO formation as well as for formation of L-citrulline (Fig. 2). These data suggest that NO synthesis is catalyzed by NO-synthase in the presence of oxygen. With an oxygen deficit (for example, in functional hypoxia associated with accelerated oxygen uptake or pathological processes following hypoxia/ischemia) the role of this NO-synthase mechanism decreases. In the following part of this review we will consider the mechanism of NO synthesis under these conditions.Fig. 1. A hypothetical model of the structure of NO-synthase and possible route of electron transport during NO-synthase reaction [69]. Domain containing NADPH, FMN, and FAD is considered cytochrome P-450 reductase. Catalytic regions include cytochrome P-450-like oxidase (heme-binding site). Regulatory domain includes calmodulin-binding site.
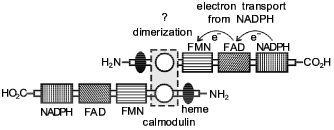
Fig. 2. Formation of NO and L-citrulline during NO-synthase reaction. One of the guanidine nitrogens of L-arginine is oxidized by molecular oxygen to NO with participation of NO-synthase. Chemical groups are indicated which are involved in modification during formation of NO and citrulline.
NITRITE-REDUCTASE COMPONENT OF THE NITRIC OXIDE CYCLE: NO SYNTHESIS IN THE ABSENCE OF OXYGEN
Activity of Nitrite-Reductase Systems in Mammals
While investigating the mechanisms of reduction of NO2- to NO in mammals, we found that the activity of nitrite-reductase systems became rather high in hemic hypoxia. Specifically, 1 h after introduction of nitrite at the dose of 5 mg/100 g body weight about 60% of total Hb in blood were present as metHb. At the same time, Hb--NO complexes account for ~10-15% of the total Hb. It is known that the normal Hb content in blood of mammals ranges from 110 to 160 g/liter or ~2·10-3 mM. This means that these 10-15% of Hb--NO complex are equal to ~2·10-4 mM. Keeping in mind that the stationary concentration of NO formed due to the functioning of NO-synthases is 1-100 nmoles or 10-7-10-9 moles/g tissue [81-83], we can deduce that this concentration is 103-104-fold lower than that which can be found in mammals as a result of the reduction of NO2- to NO. Bearing in mind that nitrite-reductase activity (1) in mammals is at least 1000-fold higher than that of NO-synthase (2), it becomes clear that the role of the former should be taken into account when studying NO synthesis:
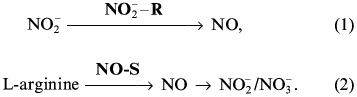
Indeed, these data make NO2- and nitrite-reductase activity in mammalian cells a part of the integral system which participates in NO synthesis (Fig. 3). The question arises: why is the nitrite-reductase activity 103-fold higher than that of NO-synthase? Below we shall try to answer this question.
Nitrite-Reductase Activity in Mammalian BloodFig. 3. NO-synthase (NO-S) and nitrite-reductase (NO2--R) components of nitric oxide cycle in mammals: 1) NO formation in NO-synthase reaction; 2) NO oxidation to NO2- and NO3- ions; 3) NO2- reduction to NO with participation of nitrite-reductase systems.
Investigation of the interaction of nitrites with hemoglobin began in 1868 [84]. Nevertheless, the question of what compounds are the immediate reducing agents for NO2- was not considered for a long time. As a rule, biochemists, toxicologists, and physicians were interested only in metHb-producing activity of NO2- but not in the nitrite-reductase activity of proteins and low-molecular-weight compounds present in blood and tissue cells of mammals. Notwithstanding the prolonged history--more than 120 years--of investigation of the action of nitrites on humans and animals, many questions still remain. Moreover, the very mechanism of hemoglobin oxidation mediated by NO2- and the corresponding reactions are not yet completely understood [85-87].
The complex character of the course of the reaction between nitrites and hemoglobin and ambiguity of the published data have made it necessary to analyze the mechanism of conversion of NO2- to NO in blood. We believe that the details of this mechanism will be supplemented and clarified in the future. But at present it seems reasonable to describe our ideas concerning the mechanism of reduction of NO2- to NO with the participation of hemoglobin in their most general terms.
Mechanisms of Reduction of NO2- to NO with the Participation of Hemoglobin and Myoglobin
After entering the blood, NO2- penetrates through erythrocyte membranes. The mechanism of penetration of NO2- through cell membranes and erythrocyte membranes in particular is not yet clearly understood, it is reasonable that a major role in this process is played by an anion exchanger known as "band 3 protein". Transport of NO2- through erythrocyte membranes occurs rapidly--we showed that even after the first 5 min following intravenous injection of NaNO2 in doses of 2-7 mg/100 g body weight rather high levels of methemoglobin (10-45%) can be found in blood. Formation of Hb--NO complexes is observed simultaneously with Hb oxidation. What is the mechanism of conversion of NO2- to NO? In experiments with hemolyzed erythrocytes we demonstrated that in the absence of oxygen the concentration of Hb--NO complexes formed in the presence of NO2- during the first hour is at least 10-fold higher than that found in the presence of oxygen. As O2 in the blood interacts predominantly with Hb, it could be deduced that hemoglobin itself plays a major role in the reduction of NO2- to NO.
In vitro experiments [1, 47, 48, 88] demonstrated that during the interaction of NO2- with deoxyHb, an oxidation-reduction reaction takes place, in the course of which deoxyHb is oxidized to metHb and NO2- is reduced into NO:
NO interacts with the reduced hemoglobin and forms stable Hb--NO complexes:
Complexes of metHb and NO are not stable [20, 89]; they readily disintegrate:
During the interaction of NO2- with oxyhemoglobin the latter is converted into its oxidized form as demonstrated by the optical absorption spectra. However, the formation of Hb--NO complexes does not take place during at least the first 3 h. Thus, we proposed the following hypothesis: during the interaction between oxyHb and NO2- a redistribution of electron density from heme Fe to oxygen and formation of O2- takes place [1, 88]:
where [Hb3+--O2-]NO2- represents oxyhemoglobin with electron density redistributed to O2-. Later on, following the loss of NO2-, this complex either converts once again into reduced hemoglobin or is oxidized to metHb as a result of removal of an electron with ligand-bound oxygen:
The formation of O2-, H2O2, and other reactive oxygen metabolites may oxidize NO to NO2- and NO3-.
Why is NO2- unable to accept electrons from oxyHb? The cause is likely to be the presence of heme-bound oxygen which prevents a contact between the donor orbital of heme Fe2+ and the acceptor orbital of NO2-. However, the bond between O2 and hemoglobin is not stable [90]:
The tertiary and quaternary structures of Hb oscillate rapidly and continuously between the oxy- and the deoxy-conformations [90]. These conformational oscillations may result in the loss of O2 and conversion of oxyhemoglobin to its deoxy form. After dissociation of O2 from Hb, NO2- near the heme iron allows the interaction between the donor 3d-electron orbital of the heme iron and a free pi*-antibonding molecular orbital of NO2-; a transfer of the 3d-electron of the heme iron to the NO2- orbital should occur. Such a possibility is in accordance with the values of the oxidation--reduction potentials of metHb/Hb = 0.17 V and NO2*-/NO = 0.37-0.38 V [90, 91]. This is followed by a breakdown of NO2- and formation of NO, the latter forming a firm bond with hemoglobin. Weakening of the bond between O2 ligand and Hb (e.g., by acidification of the incubation medium) promotes reduction of NO2- to NO. And vice versa, strengthening of the bond of the O2 ligand with hemoglobin by alkalinization largely decreases the nitrite-reductase properties of Hb. Like hemoglobin, myoglobin has a nitrite-reductase activity in its deoxy form. But, unlike Hb, myoglobin can form only R-conformers of Mb--NO complexes.
In our experiments we also revealed that the degree of the reduction of NO2- to NO depends on the activity of the metHb reductase. Indeed, 1.5-2-fold loss of metHb reductase activity in hemolyzed blood due to its storage at 4°C was accompanied by a decrease to the same extent of the content of Hb--NO complexes formed during incubation of hemolyzed blood with nitrites. This gave us grounds to suggest that an important role in reduction of NO2- to NO is played not by O2 only, but by the activity of those systems participating in Hb reduction as well. Indeed, only reduced Hb may reduce NO2- to NO. That is why there is every reason to think that the electron donor systems which take part in the process of Hb reduction play the same major role in the nitrite-reductase reaction as well. Two systems of metHb reduction are known: an enzyme system (NADH-dependent metHb reductase and NADPH-dependent metHb reductase) and a non-enzyme one (ascorbic acid and reduced glutathione). The most powerful of these systems is the NADH-dependent methemoglobin reductase [92, 93].1
1 Editor's Note: Alternative mechanism of NO generation from nitrite in animal tissues and cells through nitrite transformation to nitrous acid is considered in literature [65, 89]. The issue on correspondence of this mechanism to the mechanism of NO generation from nitrite discussed by the authors of this manuscript remains open.
Nitrite-Reductase Activity of Mitochondria and Endoplasmic Reticulum
Enzymatic reduction of NO3- and NO2- was investigated in the most detail in plants and microorganisms [94, 95]. It is known that nitrate and nitrite-reductase systems may contain pyridine nucleotides, flavoproteins, and cytochromes [94]. The same set of proteins and low-molecular-weight compounds is known to form a part of mitochondrial membranes [96]. Taking into account the similarity of the qualitative composition of mitochondrial respiratory chains and nitrate- and nitrite-reducing systems of plants and microorganisms, it could be deduced that the most probable enzymes capable of NO2- reduction are the proteins of the respiratory chain.
NO2- reduction in mitochondria. In experiments using EPR for detection it was shown that some inhibitors of the respiratory chain (e.g., rotenone) may profoundly inhibit the nitrite-reductase activity of these organelles. On the contrary, other inhibitors (cyanides) completely block the reduction of NO2- to NO and formation of paramagnetic complexes of non-heme iron with NO [1, 47]. A possibility of oxidation of the reduced mitochondrial pyridine nucleotides by NO2- through an intact respiratory chain was demonstrated by spectrofluorimetry and spectrophotometry [47]. Inhibition of the mitochondrial respiratory chain by cyanides prevented the slow decrease in the degree of reduction of pyridine nucleotides caused by electron acceptance from the respiratory chain by NO2-. It was also shown that during the interaction of NO2- with reduced NAD without intermediate carriers, the former does not oxidize this pyridine nucleotide [1, 47]. Thus, the data indicate that enzymes of the mitochondrial respiratory chain may take part in the process of reduction of NO2- to NO.
It is known that nitrite-reductases of two microorganisms, Paracoccus denitrificans (the former Micrococcus denitrificans) and Pseudomonas aeruginosa, may simultaneously perform functions of two enzymes: a nitrite-reductase and a cytochrome oxidase [97, 98]. Moreover, nitrite-reductase from Paracoccus denitrificans may interact with cytochrome c from cells of animal tissues [98]. Due to this fact, a part of the respiratory chain from cytochrome c to cytochrome oxidase was particularly interesting. The possibility of oxidation of exogenous reduced cytochrome c by NO2- through the mitochondrial respiratory chain of the liver was studied in an atmosphere of argon by optical spectrophotometry [1, 47, 48]. It was shown that in the atmosphere of argon NO2- can oxidize exogenous reduced cytochrome c through an intact respiratory chain of mitochondria. Inhibition of the respiratory chain by cyanides prevented oxidation of reduced cytochrome c by NO2- ions. If NO2- were absent from the incubation medium of mitochondria containing exogenous reduced cytochrome c, no oxidation of cytochrome c was shown in the atmosphere of argon. These data suggest that reduction of NO2- to NO in mitochondria takes place as a result of acceptance of electrons from the terminal site of the mitochondrial respiratory chain by NO2-. The most likely transport protein capable of performing the role of electron donor for NO2- is the mitochondrial cytochrome oxidase [1, 47, 48].
To clarify the suggested role of cytochrome oxidase in the reaction of reduction of NO2- to NO, experiments were performed in oxygen-free conditions on a model system containing cytochrome c, cytochrome peroxidase, and NO2-. These experiments showed that complexes of NO and cytochrome oxidase may be formed in the above system [1, 48]. These data are in agreement with the suggestion put forward above that the enzyme most likely to be capable of performing the role of electron donor for NO2- is cytochrome oxidase [1]. Our suggested mechanism of NO2- reduction to NO was independently supported by observations of Brudvig et al. [99] and Morse et al. [100], who demonstrated that NO2- can be reduced in a model system containing enzymes of the terminal region of the mitochondrial respiratory chain.
Reduction of NO2- in microsomes. Several investigations studying the possibility that NO2- can accept electrons from cytochrome P-450 of liver microsomes demonstrated that under oxygen-free conditions NO2- is capable of accepting electrons from cytochrome P-450 [101, 102]. We confirmed these results by EPR-spectroscopy. While incubating liver microsomes with NO2- under oxygen-free conditions, we detected EPR signals of complexes formed by NO and heme iron.
STRUCTURAL AND FUNCTIONAL PROPERTIES OF NO-SYNTHASE AND
NITRITE-REDUCTASE SYSTEMS
The metabolic network includes a rather wide range of different biochemical compounds and structures. But NO-synthase and nitrite-reductase systems are characterized by a rather narrow scope of properties which are defined mostly by their structure. What are the special features which characterize these systems and play a major role in their function? If we compare NO-synthase and nitrite-reductase systems with electron-transport chains (ETC) of bacteria, we can see the general similarity of their structure. Indeed, NADPH or NADH can be electron donors in NO-synthetic or nitrite-reductase reactions. Flavins--FMN or FAD--included in the structure of flavoproteins, play a role of intermediate components which participate in electron transfer from NADPH or NADH. As a matter of fact, these flavoproteins play a role of reductases of a heme-containing protein, the latter in animal cells being cytochrome P-450, cytochrome oxidase, hemoglobin, or myoglobin.
The table summarizes the main elements included in the composition of NO-synthase and nitrite-reductase systems. These data support the above-stated idea. We believe that this presentation of NO-synthase and nitrite-reductase systems in the form of distinct main blocks gives an opportunity to put well-known biochemical structures in a new light.
Basic elements of NO-synthase and nitrite-reductase systems
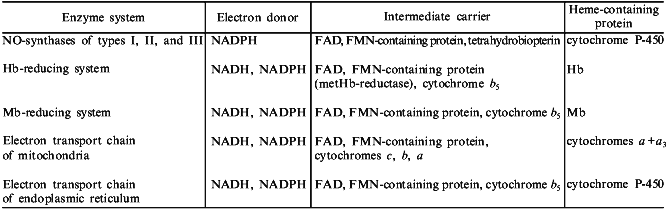
Note: Practically all enzyme systems contain either two subunits (NO-synthase
systems) or parallel electron transport chains (nitrite-reductase systems).
The mentioned data indicate that evolution of NO-synthase and nitrite-reductase systems was based on simple natural processes which included a principle of block organization of biochemical structures [103]. The basic blocks were the following: a) NADPH or NADH as an electron donor; b) flavoprotein acting as an intermediate electron carrier; and c) heme-containing protein which acts as a catalytic subunit. Therefore, NO-synthase and nitrite-reductase systems are catalytic structures which have many chemical abilities and are very stable.
Relationships between NO-synthase and nitrite-reductase activities in mammals. The analysis of nitrite-reductase systems in mammals helps to answer the following question: why is the rate of NO formation during the course of nitrite-reductase reaction at least 1000-fold higher than that of NO-synthase reaction? Indeed, NO-synthases are not present in all cells of mammals, unlike mitochondria and endoplasmic reticulum, whose electron transport chains (ETC) can participate in reduction of NO2- to NO. Besides, a substantial contribution to nitrite-reductase activity is made by Hb in blood and by Mb in muscle tissue. Keeping in mind that Hb content is approximately 250-fold higher than that of cytochromes [104], it becomes clear that the contribution of the blood nitrite-reductase systems to the total nitrite-reductase activity of mammals is maximal. This supposition is in accordance with the well-known fact that during nitrite introduction in blood R- and T-conformers of Hb--NO complexes can be found in practically all tissues [47, 48]. After washing away the blood from tissues and separation of mitochondria, one can find complexes of non-heme iron and NO. Moreover, a NO trap is not necessary for detecting such complexes (complexes of both heme and non-heme iron with NO). At the same time, it is necessary to have NO traps to detect NO resulting from the activity of NO-synthase systems; one such trap is diethylthiocarbamate [49-52, 105]. Even these qualitative data indicate that the capacity of NO-synthase systems is much less than that of nitrite-reductase systems.
In connection with that the following question arises: why is such an ineffective mechanism of NO synthesis as the NO-synthase system necessary for mammals which already have a much more powerful NO-synthetic mechanism--the nitrite-reductase system? The high NO-synthetic activity of blood Hb made it susceptible to a methemoglobin-synthetic activity of NO2-.2 Therefore, appearance (or conservation during evolution) of NO-synthase systems which limit the formation of nitrite and nitrate ensured protection of the blood pigment from oxidation and hence the organism as a whole from the toxic activity of these compounds. Besides, if an organism lacked NO-synthase reaction (i.e., lacked the mechanism for endogenous formation of NO2- and NO3- ions), its cells and the organism as a whole would be absolutely dependent on exogenous sources of nitrites and nitrates. At the same time, it is known that the best way to adapt to a variable environment is to become independent of it [106]. That is why the general line of evolution was to liberate developing organisms from the power of accidental phenomena occurring in their environment. And this, in turn, led to the formation of mechanisms of duplicating functions, although such duplication was usually carried out on the basis of numerous combinations of a limited number of initial elements [103, 106, 107]. The appearance of ETC, in particular, NO-synthase and nitrite-reductase systems possessing the optimal biochemical standard (pyridine nucleotides --> flavoproteins --> heme-containing proteins) was one of such mechanisms of duplication of functions.
2 Editor's Note: Therefore, this way of NO synthesis could result in impairment of oxygen transport in mammals.
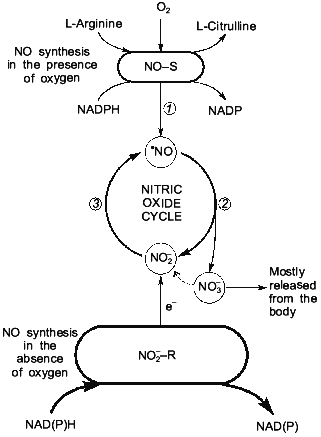
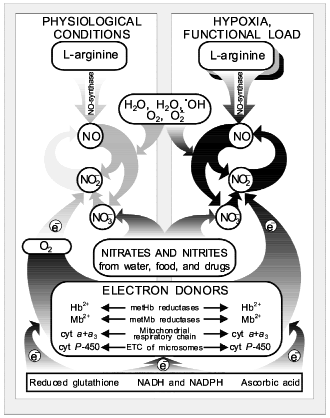
THE NITRIC OXIDE CYCLE
As shown above, the presence of the NO-synthase mechanism provides the endogenous synthesis of NO, NO2- and NO3-. And a high activity of nitrite-reductase systems (heme-containing proteins Hb, Mb, cytochrome oxidase, and cytochrome P-450) provides conditions for the chain (2) together with nitrite-reductase chain (1) function as a closed cycle, which we called the nitric oxide cycle (Fig. 4). Oxygen is needed for enzymatic oxidation of L-arginine upon participation of NO-synthases. So, the NO-synthase mechanism should be inhibited under ischemia/hypoxia. At the same time, oxygen deficiency is a factor maintaining active nitrite-reductase systems related to heme-containing proteins: Hb, Mb, cytochrome oxidase, and cytochrome P-450 [47, 48]. It has been established earlier that oxygen inhibits nitrite-reductase activity of heme-containing proteins [88] and hypoxia/ischemia and functional load activate the nitric oxide cycle [17, 47, 48]. Since only reduced forms of heme-containing proteins can reduce NO2- to NO, the role of enzymatic and nonenzymatic systems participating in the electron transfer to these heme-containing proteins must be taken into account. First they should include methemoglobin and metmyoglobin reductases as well as ETC of mitochondria and endoplasmic reticulum [92, 93]. These enzymatic systems are known to transfer electrons from NADPH or NADH through flavoproteid to heme-containing proteins. Ascorbic acid and restored glutathione should be noted among low-molecular-weight systems able to take part in restoration of heme-containing proteins [92, 93].
At present the formation of cyclic pathways in the network of metabolic reactions is thought to be universal rather than unique [108]. Moreover, cyclization of metabolic pathways is considered a mechanism providing a high degree of order and coherence of systems of biochemical reactions. Due to the presence of the cyclic pathways between individual metabolites, the increase in concentration of products capable of regeneration accelerates their transformation in the cycle and does not lead to their toxic action on cells because of accumulation of individual products. This is especially urgent in case of such highly reactive free-radical compounds as NO and NO2 are, as well as products of their metabolism. For these highly reactive compounds capable of generating free-radical chain reactions [109-111], the cycle mechanism provides not only their effective operation but also rapid removal by making them less active substances, for example NO2- and NO3-.Fig. 4. Nitric oxide cycle in mammals [47, 48]. NO generation in mammals can occur both as a result of guanidine nitrogen of L-arginine in the presence of oxygen: L-arginine --> NO (NO-synthase reaction), and on reduction of NO2- ions to NO under hypoxia: NO2- + e- --> NO (nitrite-reductase reaction).
As shown earlier, the physical and chemical properties of NO, e.g., small size and absence of charge, do not permit any isolation of this in membrane structures [47]. So, intracellular depots for NO in the form of membrane formations and intracellular structures are impossible in principle. But what regulation pathways of intracellular NO concentration remain?
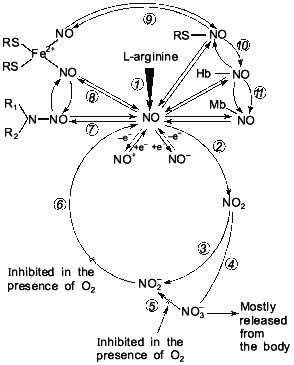
By changing the valency of the nitric atom via redox reactions, it is possible to effectively regulate the concentration of these highly reactive metabolites. This is the way which is found in the nitric circulation process (see below) in the biosphere. And in this case the same physical and chemical properties of the nitric atom, which is able to change valency from -3 to +6, are completely realized. Due to its physical and chemical properties, NO can take part in redox reactions with formation of a rather great number of intermediate compounds. Besides, NO easily forms complexes with sulfur (nitrosothiols [61-63]), with non-heme iron (Fe2+) (mono- and dinitrosyl iron complexes [50-55]), and with heme-containing proteins heme iron which is in the bivalent state [43-47]. Nitric oxides (NO and NO2) are capable of participating in free-radical nitrosylation of secondary amines with formation of N-nitrosamines [46]. All the compounds are candidates for intermediate compounds of the nitric oxide cycle (Fig. 5).
The history of science suggests that studies concerning one and the same object are, as a rule, carried out in an isolated way. In this case the results of the individual lines of investigations are either not used or completely ignored. Attempts made in order to reconstruct the object of study by one individual part of it never resulted in success [112, 113]. At the same time, sooner or later (more often it is later, when researchers have exhausted the possibilities of all round about ways and are forced by circumstances to choose the only possible direct way towards investigation), there arises such a form of interrelations between individual relative research directions when they are united for solution of the one common problem.Fig. 5. Extended variant of nitric oxide cycle in mammals [47]: 1) NO-synthase; 2-4) non-enzymatic oxidation of NO to NO2, NO2-, and NO3-; 5) Mo-containing enzymes (xanthine-oxidase, D,L-amino acid oxidase, etc.) reducing NO3- ions to NO2-; 6) heme-containing proteins reducing NO2- ions to NO; 7-11) reversible binding of NO by low- and high-molecular-weight compounds involved in EDRF formation; 12, 13) redox transformation of NO to NO+ and NO-.
In this paper we have attempted to draw some lessons from the history of studies of redox transformations of the nitrogen atom. As shown above, this history is more than 120 years old. It is characterized by many research directions related to the effect of nitro- and nitroso compounds on humans and animals and by analysis of the role of these compounds in regulatory, pharmacological, toxic, and cancerogenic actions. During the last decade NO-synthase and nitrite-reductase reactions in mammals were studied separately, despite their structural and functional similarity. The popularity of the NO molecule in biological systems drew back the products of transformation of this molecule (NO2- and NO3-) and their metabolism in animals. At the same time, some attempts are now made to generalize the evidence on the role of NO in biological systems without taking into account metabolism of nitrogen oxide itself and the compounds that accompany the NO-synthase reaction. Taking into account that it is practically impossible to reconstruct the research object by one individual part of it [112, 113], we tried several times to show the qualitative shift in the research process to be probable only on the way of a complex study of NO-synthase and nitrite-reductase processes. As a result of analysis of data in the literature and our own evidence, we were the first to show the electron-donor systems containing NADH, NADPH, flavoproteids and reduced heme-containing proteins, which are in the deoxy-form, to possess nitrite-reductase activity and to be capable of closing the chain of metabolic transformations L-arginine --> NO --> NO2-/NO3- to the cycle that we have called the nitric oxide cycle [47, 48]. Most likely, this cycle arose as a result of the opposing development of nitrite-reductase and NO-synthase components. Thus, on the basis of the data obtained by us and analysis of evidence in the literature we formulated a conception of the nitric oxide cycle, new in principle, which allowed us to come up to analysis of the interrelation between diatomic NO molecule, NO2-, and the evolution of energetic systems of cells.
We believe that the aim of the conception suggested by us and substantiation of the nitric oxide cycle's existence in mammals will be justified if they serve as a basis for generalizing much evidence on the role of NO in biological systems available at present as well as for analyzing them in a positive and critical manner. If our concept is not accepted by some researchers, it makes sense to remember the words of A. Whitehead which are constantly confirmed by the history of science -- "collision of theories is not distressing, but favorable because it opens new perspectives".
REFERENCES
1.Reutov, V. P., Sorokina, E. G., Okhotin, V. E., and
Kositsyn, N. S. (1997) Cyclic Transformations of Nitric Oxide in
Mammals [in Russian], Nauka, Moscow.
2.Bashkatova, V. G., Vitskova, G. Yu., Narkevich, V.
B., Vanin, A. F., Mikoyan, V. D., Kubrina, L. N., and Rayevsky, K. S.
(1996) Neirokhimiya, 13, 110-115.
3.Bashkatova, V. G., Mikoyan, V. D., Kosacheva, E.
S., Kubrina, L. N., Vanin, A. F., and Rayevsky, K. S. (1996) Dokl.
Ros. Akad. Nauk, 348, 119-121.
4.Kalinichenko, S. G., Okhotin, V. E., and Motavkin,
P. A. (1997) Tsitologiya, 39, 161-165.
5.Akaike, T., Noguchi, Y., Ijiri, S., Setiguchi, K.,
Suga, M., Zheng, Y. M., Dietzschold, B., and Maeda, H. (1996) Proc.
Natl. Acad. Sci. USA, Microbiology, 93,
2448-2453.
6.Bastian, N. R., Foster, M. J. P., and Pope, J. C.
(1995) BioFactors, 5, 5-10.
7.Maeda, H., Akaike, T., Yoshida, M., and Suga, M.
(1994) J. Leukocyte Biol., 56, 588-592.
8.Ilnitsky, A. P., Reutov, V. P., Ryzhova, N. I.,
Kolpakova, A. S., Deryagina, V. P., Nekrasova, E. N., Savluchinskaya,
L. A., and Travkin, A. G. (1997) Exp. Oncol., 19,
1-9.
9.Vanin, A. F. (1991) FEBS Lett., 289,
1-3.
10.Berdiev, U. B., Reutov, V. P., Vishnevsky, E. S.,
Sheksheev, E. M., and Kayushin, L. P. (1990) Biofizika,
35, 382-383.
11.Vanin, A. F. (1993) Biofizika, 38,
751-761.
12.Gorbunov, N. V., and Avrova, N. F. (1995)
Neurokhimiya, 12, 9-18.
13.Gurin, A. V. (1997) Uspekhi Fiziol. Nauk,
28, 53-60.
14.D'yakonova, T. L., and Reutov, V. P. (1994)
Vopr. Med. Khim., 6, 20-25.
15.Kudryavtsev, M. E., Dmitrieva, O. N., and
Kuropteva, Z. V. (1996) Izvestiya Akad. Nauk. Ser.
Biol., 4, 453-459.
16.Pivovarov, A. S., and Ekhido-Villareal, U. (1995)
Zh. Vysshei Nervn. Deyat. Neirofiziol., 45,
558-564.
17.Rayevsky, K. S. (1997) Byul. Eksp. Biol.
Med., 5, 484-490.
18.Urazaev, A. Kh., Magsumov, S. T., Naumenko, N.
V., and Poletaev, G. I. (1995) Neirokhimiya, 12,
46-50.
19.Urazaev, A. Kh., Magsumov, S. T., Naumenko, N.
V., and Poletaev, G. I. (1995) Neirokhimiya, 13,
52-55.
20.Henry, Y., Guissani, A., and Ducastel, B. (1997)
Nitric Oxide Research from Chemistry to Biology: EPR
Spectroscopy of Nitrosylated Compounds, Springer-Verlag, New
York.
21.Avdonin, P. V., and Tkachuk, V. A. (1994)
Receptors and Intracellular Calcium [in Russian], Nauka,
Moscow.
22.Reutov, V. P., and Orlov, S. N. (1993) Fiziol.
Cheloveka, 19, 124-137.
23.Garthwaite, J. (1991) Trends Neurosci.,
14, 60-67.
24.Gimelbrand, A. A. (1994) Biochemistry
(Moscow), 59, 748-749 (Russ.).
25.Galione, A. (1992) Trends Pharmacol.
Sci., 13, 304-306.
26.Brune, B., and Lapetina, E. (1989) J. Biol.
Chem., 264, 8455-8458.
27.Lipton, S. A., Choi, Y.-B., and Pan, Zh.-H.
(1993) Nature, 364, 626-632.
28.Dawson, V. L., Dawson, T. M., London, E. D.,
Bredt, D. S., and Snyder, S. H. (1991) Proc. Natl. Acad. Sci.
USA, 88, 6368-6371.
29.Dawson, V. L., Dawson, T. M., Bartley, D. A.,
Uhl, G. R., and Snyder, S. H. (1993) J. Neurosci.,
13, 2651-2661.
30.Knowles, R. G., and Moncada, S. (1992) Trends
Biochem. Sci., 17, 399-402.
31.Knowles, R. G., Palacios, M., Palmer, R. M. J.,
and Moncada, S. (1989) Proc. Natl. Acad. Sci. USA,
89, 5159-5162.
32.Moncada, S., Palmer, R. M. J., and Higgs, E. A.
(1992) Pharmacol. Rev., 43, 109-142.
33.Palmer, R. M. J., Ashton, D. S., and Moncada, S.
(1988) Nature, 333, 664-666.
34.Palmer, R. M. J., Rees, D. D., Ashton, D. S., and
Moncada, S. (1988) Biochem. Biophys. Res. Commun.,
153, 1251-1256.
35.Palmer, R. M. J., Ferrige, A. G., and Moncada, S.
(1987) Nature, 327, 524-526.
36.Snyder, S. H. (1992) Science, 257,
494-496.
37.Vanin, A. F. (1995) Biochemistry (Moscow),
60, 225-229 (Russ.).
38.Vanin, A. F., Blumenfeld, L. A., and Chetverikov,
A. G. (1967) Biofizika, 12, 829-838.
39.Vanin, A. F., and Burbaev, D. Sh. (1976) Zh.
Vsesoyuz. Khim. Obshch. im. D. I. Mendeleeva, 21,
672-684.
40.Kuropteva, Z. V., and Pastushenko, O. N. (1985)
Dokl. Akad. Nauk SSSR, 281, 189-192.
41.Reutov, V. P., Kayushin, L. P., and Sorokina, E.
G. (1994) Fiziol. Cheloveka, 20, 165-174.
42.Pulatova, M. K., Rikhireva, G. T., and Kuropteva,
Z. V. (1989) Electron Paramagnetic Resonance in Molecular
Radiobiology [in Russian], Energoatomizdat, Moscow.
43.Saprin, A. N., Kozlova, L. E., Shabalkin, V. A.,
Kruglyakova, K. E., and Emanuel, N. M. (1969) Izvestiya Akad. Nauk
SSSR. Ser. Biol., 6, 887-892.
44.Saprin, A. N., Shabalkin, V. A., Kozlova, V. A.,
Kruglyakova, K. E., and Emanuel, N. M. (1968) Dokl. Akad. Nauk
SSSR, 181, 1520-1523.
45.Saprin, A. N., and Shulyakovskaya, T. S. (1969)
Dokl. Akad. Nauk SSSR, 189, 889-891.
46.Rubenchik, B. L., Osinkovskaya, N. D.,
Mikhailenko, V. M., Furman, M. A., and Boim, T. M. (1990) J.
Environ. Pathol. Toxicol. Oncol., 10, 290-296.
47.Reutov, V. P. (1995) Uspekhi Biol. Khim.,
35, 189-228.
48.Reutov, V. P., Kayushin, L. P., and Sorokina, E.
G. (1994) Vopr. Med. Khim., 40, 31-35.
49.Vanin, A. F., Kubrina, L. N., and Mordvintcev, P.
I. (1988) Dokl. Akad. Nauk SSSR, 301, 490-493.
50.Vanin, A. F., and Malenkova, I. V. (1996)
Biochemistry (Moscow), 61, 505-513 (Russ.).
51.Vanin, A. F., Malenkova, I. V., Mordvintcev, P.
I., and Mulsh, A. F. (1993) Biochemistry (Moscow), 58,
1094-1103 (Russ.).
52.Vanin, A. F., Manukhina, E. B., Lapshin, A. V.,
and Meerson, F. Z. (1993) Byul. Eksp. Biol. Med.,
116, 142-144.
53.Manukhina, E. B., Azamatov, Z. Z., Malysheva, E.
V., and Malyshev, I. Yu. (1996) Fiziol. Zh. im. I. M. Sechenova,
82, 60-65.
54.Manukhina, E. B., Malyshev, I. Yu., Mikoyan, V.
D., Kubrina, L. N., and Vanin, A. F. (1996) Byul. Eksp. Biol.
Med., 5, 520-523.
55.Mikoyan, V. D., Kubrina, L. N., and Vanin, A. F.
(1996) Biochemistry (Moscow), 61, 1182-1188 (Russ.).
56.Okhotin, V. E., and Kupriyanov, V. V. (1996)
Morfologiya, 110, 17-22.
57.Malyshev, I. Yu., Malugin, A. V., Manukhina, E.
B., Larionov, N. P., Malenyuk, E. V., Malysheva, E. V., Mikoyan, V. D.,
and Vanin, A. F. (1996) Physiol. Res., 45, 267-272.
58.Maeda, H., Akaike, T., Yoshida, M., Sato, K., and
Noguchi, Y. (1995) in The Role of Nitric Oxide in Physiology
and Pathophysiology (Koprowski, H., and Maeda, H., eds.)
Springer-Verlag, Berlin, Heidelberg, pp. 37-50.
59.Manukhina, E. B., Lapshin, A. V., Mashkina, S.
Yu., Meerson, F. Z., Mikoyan, V. D., Kubrina, L. N., and Vanin, A. F.
(1995) Byul. Eksp. Biol. Med., 11, 495-497.
60.Vanin, A. F., Stukan, R. A., and Manukhina, E. B.
(1996) Biochim. Biophys. Acta, 1295, 5-12.
61.Ignarro, L. J., Lipton, H., Edwards, J. C.,
Baricos, W. H., Hyman, A. L., Kadowitz, P. J., and Gruetter, C. A.
(1981) J. Pharmacol. Exp. Ther., 218, 739-749.
62.Ignarro, L. J. (1990) Annu. Rev. Pharmacol.
Toxicol., 30, 535-560.
63.Ignarro, L. J., Buga, G. M., Wood, K. S., Byrus,
R. E., and Chandhuri, G. (1987) Proc. Natl. Acad. Sci.
USA, 84, 9265-9269.
64.Zweier, J. L., Wang, P. A., and Kuppusamy, P.
(1995) J. Biol. Chem., 270, 304-307.
65.Zweier, J. L., Wang, P., Samouilov, A., and
Kuppusamy, P. (1995) Nature Medicine, 1, 804-809.
66.Abu-Soud, H. M., and Stuehr, D. J. (1993)
Proc. Natl. Acad. Sci. USA, 90, 10769-10772.
67.Nathan, C. (1991) Res. Immunol.,
142, 600-602.
68.Nathan, C. (1995) in The Role of Nitric Oxide
in Physiology and Pathophysiology (Koprowski, H., and Maeda, H.,
eds.) Springer-Verlag, Berlin, Heidelberg, pp. 75-86.
70.Bredt, D. S., Hwang, P. M., Glatt, C. E.,
Lowenstein, C., Reed, R. R., and Snyder, S. H. (1991) Nature,
351, 714-718.
71.Fazeli, M. S. (1992) Trends Neurosci.,
15, 115-117.
72.Hammer, B., Parker, W. D., and Bennet, J. P., Jr.
(1993) Neuro Report, 5, 72-74.
73.Nakane, M., Schmidt, H. H. H. W., Pollock, J. S.,
Pollock, J. S., Forstermann, U., and Murad, F. (1993) FEBS
Lett., 316, 175-180.
74.Stuehr, D. J., Cho, H. J., Kwon, N. S., Weise,
M., and Nathan, C. F. (1991) Proc. Natl. Acad Sci. USA,
88, 7773-7777.
75.Stuehr, D. J., and Marletta, M. A. (1987) J.
Immunol., 139, 518-525.
76.Chartrain, N. A., Geller, D. A., Koty, P. P.,
Sitrin, N. F., Nussler, A. K., Hoffman, E. P., Billiar, T. R., and
Hutchinson, N. I. (1994) J. Biol. Chem., 269,
6765-6772.
77.Lyons, C. R., Orloff, G. J., and Cunningham, J.
M. (1992) J. Biol. Chem., 267, 6370-6374.
78.Xie, Q.-W., Cho, H. I., Calaycay, J., Mumford, R.
A., Swiderek, K. M., Lee, T. D., Ding, A., Troso, T., and Nathan, C.
(1992) Science, 256, 225-228.
79.Lowenstein, C., Glatt, C. S., Bredt, D. S., and
Snyder, S. H. (1992) Proc. Natl. Acad. Sci. USA,
89, 6711-6715.
80.Janssens, S. P., Shimouchi, A., Quertemous, T.,
Bloch, D. B., and Bloch, K. D. (1992) J. Biol. Chem.,
267, 14519-14522.
81.Garthwaite, J. (1990) in Nitric Oxide from
L-Arginine: a Bioregulatory System (Moncada, S., and Higgs, E. A.,
eds.) Excerpta Medica, Amsterdam, pp. 115-138.
82.Garthwaite, J., Charles, S. L., and
Chess-Williams, R. (1988) Nature, 336, 385-388.
83.Vanin, A. F., Kubrina, L. N., and Mordvintcev, P.
I. (1988) Dokl. Akad. Nauk SSSR, 301, 490-492.
84.Gamge, A. (1868) Phil. Pr. Roy. Soc. L.,
158, 589-599.
85.Ebert, B., Jung, F., and Lassmann, G. (1982)
Stud. Biophys., 89, 65-69.
86.Mansouri, A. (1981) Experientia,
37, 1199-1201.
87.Doyle, M. P., Pickering, R. A., and Dyketra, R.
L. (1982) Biochem. Biophys. Res. Commun., 105,
127-132.
88.Reutov, V. P., Azhipa, Ya. I., and Kayushin, L.
P. (1983) Izv. Akad. Nauk SSSR. Ser. Biol., 3,
408-418.
89.Marshall, W., and Marshall, C. R. (1945) J.
Biol. Chem., 158, 187-208.
90.Volkenshtein, M. V. (1975) Molecular
Biophysics [in Russian], Nauka, Moscow.
91.Dickerson, R. E., and Timkovich, R. (1975) in
The Enzyme V.XI. Oxidation-Reduction. Pt. A, Academic Press, New
York, pp. 397-549.
92.Kushakovsky, M. S. (1968) Clinical Forms of
Hemoglobin Damage [in Russian], Meditsina, Leningrad.
93.Topunov, A. F., and Golubeva, L. I. (1989)
Usp. Biol. Khim., 30, 239-252.
94.L'vov, N. P. (1989) Molybdenum in Nitrogen
Assimilation in Plants and Microorganisms [in Russian], Nauka,
Moscow.
95.Zavarzin, G. A. (1972) Litotrophic
Microorganisms [in Russian], Nauka, Moscow.
96.Zvyagil'skaya, R. A., and Kotel'nikova, A. V.
(1989) Usp. Biol. Khim., 30, 161-189.
97.Kristjansson, J. K., and Hollocher, T. C. (1981)
Curr. Microbiol., 6, 247-251.
98.Smith, L., Newton, N., and Scholes, P. B. (1966)
Hemes and Hemoproteins (Chance, B., Estabrook, R. W., and
Yonetani, T., eds.) Academic Press, New York, pp. 395-403.
99.Brudvig, G. W., Stevens, T. H., and Chan, S. J.
(1980) Biochemistry, 19, 5275-5285.
100.Morse, R. H., and Chan, S. J. (1980) J.
Biol. Chem., 255, 7876-7882.
101.Raikhman, L. M., and Annaev, B. (1972) in
Study of Free Radical States in Relation with Their Role in
Regulation of Biological Processes [in Russian], Institute of
Biological Physics, USSR Academy of Sciences, Pushchino, pp. 11-22.
102.Duthu, G. S., and Shertzer, H. G. (1979)
Drug. Metab. Dispoit. Biol. Fate Chem., 7, 263-269.
103.Ivanitsky, G. R., Esipova, N. G., Abagyan, R.
A., and Shnoll, S. E. (1985) Biofizika, 30, 418-421.
104.Margelis, L. (1983) Role of Symbiosis in
Cell Evolution [Russian translation], Mir, Moscow.
105.Lobysheva, I. I., Serezhenkov, V. A., Stukan,
R. A., Bouman, M. K., and Vanin, A. F. (1997) Biochemistry
(Moscow), 62, 934-943 (Russ.).
106.Gomazkov, O. A. (1992) Functional
Biochemistry of Regulatory Peptides [in Russian], Nauka,
Moscow.
107.Nefedov, V. P., Yasaitis, A. A., and
Novosel'tsev, V. N. (1991) Homeostasis on Different Levels of
Organization of Biosystems [in Russian], Nauka, Moscow.
108.Malygin, A. G. (1984) Symmetry of Network of
Metabolism Reactions [in Russian], Nauka, Moscow.
109.Shugalei, I. V., Lopatina, N. N., Tselinsky, I.
V., and Panasyuk, S. L. (1986) Zh. Obshch. Khim., 56,
188-192.
110.Shugalei, I. V., Lopatina, N. N., Tselinsky, I.
V., and Panasyuk, S. L. (1986) Zh. Obshch. Khim., 56,
192-197.
111.Shugalei, I. V., and Tselinsky, I. V. (1992)
Zh. Obshch. Khim., 62, 155-158.
112.Rozov, M. A. (1981) Voprosy Filosofii,
8, 143-154.
113.Maidanov, A. S. (1993) Discovery Skill:
Methodology and Logic of Scientific Creation [in Russian], Repro,
Moscow.