
|
REVIEW: Properties, Functions, and Secretion of Human MyeloperoxidaseJ. ArnholdInstitut für Medizinische Physik und Biophysik, Universität Leipzig, Liebigstr. 27, D-04103 Leipzig, Germany; fax: +49-341-9715709; E-mail: arnj@medizin.uni-leipzig.de
|
Received April 30, 2003
The heme-containing protein myeloperoxidase is released from stimulated polymorphonuclear leukocytes at sites of inflammation. It is involved in the generation of reactive oxygen and nitrogen species and tissue damage. The general properties and functional aspects of this enzyme are reviewed. Special attention is given to luminescence methods for investigating the release of myeloperoxidase from stimulated cells.
KEY WORDS: myeloperoxidase, polymorphonuclear leukocytes, reactive oxygen species, hypochlorous acid, luminescence
Polymorphonuclear leukocytes (PMNs, also called neutrophils) are the first cell type in human beings that are activated in host immune defense against infection. These cells migrate, driven by chemotactic gradients, to inflammatory loci, where they recognize and phagocytize bacteria and other extrinsic microorganisms. Foreign pathogens are exposed to an arsenal of hydrolytic enzymes and bactericidal proteins prestored in granules as well as to newly generated reactive oxygen species (ROS) [1-3].
PMNs are equipped with four types of granules containing different proteins and enzymes [4, 5]. The granule content is gradually released either into the formed phagosomes or into the extracellular space upon activation of cells. Increase in intracellular calcium triggers the degranulation. For the release of the content of secretory vesicles and tertiary granules, only a moderate increase in Ca2+ to 0.25 µM is quite sufficient. These types of granules contain primarily albumin, gelatinase, collagenase, and other proteins. These proteins and enzymes are helpful to smooth the way for PMNs through the closely packed tissue material to the inflammatory loci.
For the release of the content from secondary and azurophilic granules, a much higher increase in intracellular calcium to 0.7 µM is required. These enzymes and proteins are directed most of all against foreign pathogens. There are special enzymes that cleave components of the bacterial wall.
Azurophilic granules of PMNs contain in huge amount of a unique protein, the heme-containing enzyme myeloperoxidase. Together with the membranous NADPH oxidase, myeloperoxidase is involved in the formation of reactive oxygen species and oxidation of biological material. In stimulated PMNs, NADPH oxidase reduces molecular oxygen to superoxide anion radical [6, 7]. This species and its dismutation product hydrogen peroxide are substrates for myeloperoxidase.
This review gives an overview on the important properties of myeloperoxidase and summarizes the contribution of this enzyme to host defense and tissue damage. Special attention will be focused on chemiluminescence methods to investigate the release of myeloperoxidase from phagocytes.
PROPERTIES OF MYELOPEROXIDASE
Enzyme structure. Myeloperoxidase (MPO; donor, hydrogen peroxide oxidoreductase, EC 1.11.1.7) is unique to neutrophils and monocytes. However, monocytes contain only one third of the MPO found in PMNs. Eosinophilic granulocytes contain a related enzyme, eosinophil peroxidase [8]. Both peroxidases belong to the mammalian peroxidase superfamily and share an overall homology of 69.8% at the amino-acid sequence level [9].
MPO is a strongly cationic glycosylated protein and has a molecular weight of 144 kD. It consists of each two identical dimers linked by a disulfide bridge [10]. Each dimer is made up of a light and a heavy subunit, where the latter contains a protoporphyrin IX group with a central iron ion. Both heme groups of MPO are functionally identical [11].
The three-dimensional X-ray crystal structure of human MPO has been recently obtained with a resolution of 1.8 Å [12]. The hemes are joined with the apoprotein by two ester linkages and one sulfonium ion linkage [12]. This threefold linkage of heme is unique compared to other heme proteins. It makes the porphyrin ring slightly curved and is discussed as the reason for the red shift of the Soret band to 428 nm in MPO. Different binding sites for halides have been identified.
Formation of strong oxidants by MPO. Myeloperoxidase is able to form a wide variety of oxidants. Furthermore, activated states of the enzyme (most of all compound I, but also compound II) are also able to oxidize different substrates. In the native enzyme, the heme iron of MPO is in the ferric state.
Native MPO binds either superoxide anion radicals or hydrogen peroxide. In the first case compound III of the enzyme is formed, which is apparently involved in the hydroxylation of aromatic substrates [13].
Hydrogen peroxide is reduced to water by native MPO upon the formation of compound I of the enzyme. Alternatively, different organic hydroperoxides [14] as well as hypochlorous acid in chloride-free medium [15, 16] are also able to oxidize the native MPO to compound I. The heme iron is in the ferryl state (Fe4+) in compound I and a further oxidizing equivalent is present in the form of a porphyryl radical. Thus, compound I can be regarded as a ferryl pi-cation radical species where an oxygen atom is coupled by a double bond to the iron [17].
Compound I of MPO is reduced to the native enzyme either by abstracting two electrons from (pseudo)halides or by two one-electron steps via the formation of compound II. An overview about the halogenation and peroxidase cycle of MPO is schematically given in Fig. 1. In the first case, (pseudo)halides are oxidized to (pseudo)hypohalous acids. The myeloperoxidase-catalyzed formation of hypochlorous acid and hypothiocyanate is especially important under physiological conditions [18].
Many different substrates are known to be oxidized by compound I (and also at lower rates by compound II) by abstracting only one electron on formation of radical products. These substrates include tyrosine, tryptophan, sulfhydryls, phenol and indole derivatives, nitrite, hydrogen peroxide, xenobiotics, and others [19-21]. Numerous radical products, (pseudo)hypohalous acids and further products are involved in damage of biological macromolecules and tissue degradation.Fig. 1. Halogenation and peroxidase cycles of myeloperoxidase. Both cycles utilize hydrogen peroxide to oxidize the native enzyme to compound I. Por denotes porphyrin. AH2 and ·AH represent substrates being oxidized and the formed radical product, respectively. Instead of Cl- other (pseudo)halides such as Br-, I-, and SCN- will also be oxidized by compound I. Other pathways of myeloperoxidase are only denoted.
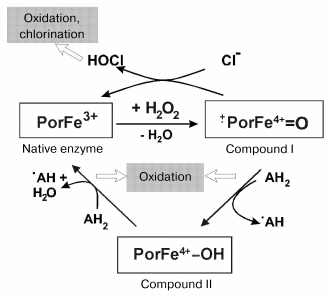
Other reactions of ferric myeloperoxidase are the reduction to the ferrous state, the reaction with nitric monoxide or superoxide anion radicals. These pathways are denoted in Fig. 1. Details can be found elsewhere [13, 22, 23].
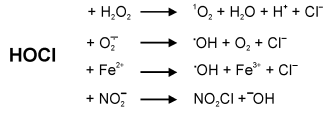
Formation of further reactive oxygen species by MPO. The formation of hypochlorous acid by myeloperoxidase is well described [24, 25]. Some reactions of hypochlorous acid lead to further reactive oxygen species with high potential for tissue damage. Hypochlorous acid reacts with hydrogen peroxide on the formation of singlet oxygen [26]. Hydroxyl radicals are not only formed as the result of the Fenton reaction. They are also derived from hypochlorous acid either by their reaction with superoxide anion radicals [27, 28] or Fe(II) [29, 30]. Hypochlorous acid also reacts with nitrite to yield the powerful chlorinating and nitrating compound NO2Cl [31]. An overview of these reactions of hypochlorous acid is given in Fig. 2.
Redox properties. Many reactions catalyzed by myeloperoxidase are redox reactions. The standard reduction potential of the redox couple compound I/native MPO has recently been determined by titration with hydrogen peroxide using stopped flow techniques to be 1.16 V at pH 7.0 [32]. The couple compound II/native MPO has a lower value for the standard reduction potential of 0.97 V at pH 7.0 [33]. Consequently, the standard reduction potential of the couple compound I/compound II of myeloperoxidase was calculated to be 1.35 V [33]. The latter value is one of the highest reduction potentials found in cellular systems. These data reflect also big differences in thermodynamic properties between compounds I and II of MPO. Thus, myeloperoxidase is able to oxidize various biological molecules with significant rates.Fig. 2. Formation of reactive oxygen and nitrogen species derived from the myeloperoxidase product hypochlorous acid.
FUNCTIONS OF MYELOPEROXIDASE
Reactions of hypochlorous acid. Oxidative equivalents formed by MPO are involved in numerous processes of tissue damage. Hypochlorous acid is known to oxidize at a significant rate sulfhydryl and thioether groups of proteins [34, 35]. It chlorinates amino groups to chloramines [36, 37]. Because of its strongly cationic properties, MPO is known to be easily attached to negatively charged biological membranes [38]. Thus, reactions with unsaturated bonds of different phospholipids are also quite possible. The formation of chlorohydrins as well as lysophospholipids in unsaturated phosphatidylcholines by hypochlorous acid and by the MPO-H2O2-Cl- system has been recently described [39-41]. MPO products are also involved in initiation of lipid peroxidation. Peroxidation by hypochlorous acid is favored by the presence of hydroperoxides previously accumulated in lipid material [42, 43]. The one-electron oxidation of different substrates by complex I of MPO causes radical products such as tyrosyl radical, which is also known to initiate lipid peroxidation processes [44, 45].
Attachment of myeloperoxidase to membranes. Plasma levels of free MPO are often elevated in patients during inflammatory conditions [46, 47]. This strongly cationic protein binds to the negatively charged endothelial plasma membrane, whereby this binding depends on the presence of heparin/heparan-containing glycosaminoglycans [48, 49]. Moreover, cell-bound myeloperoxidase rapidly transcytoses the intact endothelium and localizes at the basolateral site of the endothelium closely associated with interstitial matrix proteins such as fibronectin [49].
Protein nitration by myeloperoxidase. Nitration of free and protein-bound tyrosine correlates well with myeloperoxidase activity under inflammatory conditions [50, 51]. Compound I of MPO is able to oxidize nitrite to the nitrating species nitrogen dioxide (NO2) at a significant rate [21]. In inflammatory models, the immunoreactivity of MPO strongly co-localizes with the formation of nitrotyrosine in subendothelial and epithelial tissue regions [52].
Modulation of the vessel tonus. Myeloperoxidase impairs NO-dependent blood vessel relaxation and guanylate cyclase activation in an inflammatory model by regulating the availability of nitric oxide [53]. This effect is favored by endothelial localization of secreted MPO [52] and the high rate of NO oxidation by radical products of MPO catalysis [22, 23, 53].
Termination of PMN responses. Myeloperoxidase also modulates a variety of aspects of the inflammatory response. It is assumed that the MPO-H2O2-halide system inactivates some of the secreted granule contents, decreases the binding of formylated peptides to chemotactic receptors, and influences other functions in stimulated PMNs [54]. Thus, myeloperoxidase contributes to physiological feedback of recruitment of PMNs. It contributes to the termination of the influx of PMNs in inflammatory loci. MPO-deficient PMNs usually exhibit a stronger and more prolonged respiratory burst [55, 56].
Myeloperoxidase and pathologies. Myeloperoxidase is assumed to be involved in the pathology of different diseases such as atherosclerosis, cancer, multiple sclerosis, and Alzheimer's disease [57-60]. MPO has been found in arteriosclerotic plaques [61, 62].
ASSESSMENT OF THE RELEASE OF MYELOPEROXIDASE FROM STIMULATED
PHAGOCYTES
Marker enzymes for the degranulation of azurophilic granules. Azurophilic granules contain in addition to myeloperoxidase a variety of aggressive enzymes including elastase, cathepsins, beta-glucuronidase, beta-galactosidase, lysozyme, and others. Elastase is often used as a marker enzyme to follow the degranulation of azurophilic granules. Elastase can be easily detected in the supernatant of cells by the release of p-nitroaniline from MeO-Suc-Ala-Ala-Pro-Val-p-nitroanilide that is cleaved by elastase [63].
Another marker enzymes for the degranulation of azurophilic granules is beta-glucuronidase, which is determined using 4-nitrophenyl-beta-D-glucopyranoside uronic acid as a specific substrate.
Chemiluminescence approaches for the detection of myeloperoxidase. Chemiluminescence approaches have been widely used to detect the activation of PMNs. The main advantages of these methods are the use of a relatively low number of cells and the possibility of getting time resolved responses. Any events are immediately detected using luminescent methods. Although a low-level spontaneous light emission was originally observed in stimulated PMNs [64], the light emission in cell suspensions is mainly detected in the presence of special chemical systems that emit intense light upon oxidation. Primarily, 5-amino-2,3-dihydro-1,4-phthalazinedione (luminol) has been used as “light amplifier” in suspensions of stimulated neutrophils.
Luminol is oxidized by a variety of one- and two-electron oxidants including Fe(II) complexes, phenoxyl radicals, hypochlorous acid, and others [65-67]. Hydrogen peroxide reacts with oxidized luminol to form an endoperoxide that is finally converted to 3-aminophthalate formed in the excited state [67, 68]. The latter compound emits blue light with a wavelength maximum at 425 nm. Because of many factors influencing the light emission in the luminol system, it is hardly to answer which reactive oxygen species or oxidizing systems are responsible for the light emission of luminol in stimulated neutrophils. Moreover, luminol penetrates easily into cells. Thus, the use of impermeable inhibitors influences only partially the luminescence response in stimulated PMNs.
Because the extracellular luminescence of stimulated PMNs can be completely inhibited by scavengers of hypochlorous acid, it is believed that hypochlorous acid produced by the myeloperoxidase system contributes to the luminol luminescence in PMN suspensions. Such results have been obtained using the chemotactic tripeptide fMet-Leu-Phe as cell stimulator [69, 70]. In this case, a first maximum of light emission occurs during the first three-four minutes after stimulation that is produced mainly extracellularly. Moreover, the co-stimulation of PMNs by fMet-Leu-Phe and cytochalasin B amplifies drastically the luminescence signal. An enhancement of degranulation of azurophilic granules is known to occur under the influence of cytochalasin B.
Other authors favor the formation of peroxynitrite by PMNs as the main events that cause the luminol oxidation [71]. PMNs are known to produce peroxynitrite under certain circumstances [72].
Pholasin chemiluminescence and MPO release. Pholasin, the photoprotein of the common piddock Pholas dactylus, emits intense luminescence upon oxidation [73]. This emission is so strong that it is even possible to detect the oxidative burst in a single phagocytosing cell [74]. Pholasin is a glycosylated 34 kD protein consisting of 226 amino acid residues without any chromophoric group [75]. The light-emitting group of pholasin is still unknown.
A selected example for the pholasin luminescence of fMet-Leu-Phe stimulated PMNs is given in Fig. 3. The light emission in the pholasin system is about two orders of magnitude enhanced in comparison to luminol at the same cell concentration. Because of its size, pholasin does not penetrate into the cells. Thus, only extracellular events can be detected by pholasin.
Careful investigation on pholasin luminescence in artificial systems revealed that superoxide anion radicals as well as activated states of heme peroxidases such as horseradish peroxidase or myeloperoxidase are able to oxidize pholasin during light emission [76]. On the other hand, hydrogen peroxide is unable to interact with pholasin at micromolar concentrations [77]. The reagent hypochlorous acid was also found to oxidize pholasin [76, 77]. However, hypochlorous acid produced by the MPO-H2O2-Cl- system contributed only to a small extent to pholasin luminescence as shown by inhibition experiments with taurine or methionine. Apparently compound I and perhaps also compound II of MPO are involved in Pholasin oxidation, but not hypochlorous acid formed by the MPO-H2O2-Cl- system [76].Fig. 3. Luminol- (1) and pholasin-dependent (2) luminescence of polymorphonuclear leukocytes stimulated with the chemotactic tripeptide fMet-Leu-Phe. A 25,000-cell suspension in Hanks' balanced salt solution was incubated with either luminol (5*10-5 M final concentration) or pholasin (7*10-8 M final concentration) for 5 min at 37°C and then stimulated with fMet-Leu-Phe (10-6 M). The total volume was 250 µl. The chemiluminescence was measured with a MicroLumat LB 96 P luminometer (EG&G Berthold, Germany).
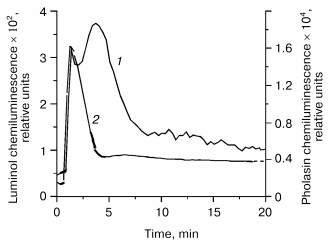
The pholasin luminescence of PMNs stimulated by different soluble stimuli was efficiently quenched by superoxide dismutase and by the myeloperoxidase inhibitor potassium cyanide, while the HOCl scavenger taurine was without any effect [76, 78]. Thus, primarily superoxide anion radicals as well as myeloperoxidase contributes to pholasin oxidation in suspensions of stimulated neutrophils. On this basis, it was possible to obtain a time-resolved profile for the generation of superoxide anion radicals as well as for the MPO activity. In cells stimulated by fMet-Leu-Phe alone, there was only a small contribution of MPO to pholasin luminescence. Here, superoxide anion radicals generated extracellularly by the NADPH oxidase were the dominating species. In contrast, a considerably higher contribution of myeloperoxidase to pholasin luminescence was observed in PMNs stimulated with a mixture of fMet-Leu-Phe and cytochalasin B. Similarly, the use of the phorbol myristate acetate (PMA), which activates directly protein kinase C, revealed also that both superoxide anion radicals and myeloperoxidase are involved in light emission of pholasin. During the first three minutes, the contribution of superoxide anion radical dominates, whereas the contribution of myeloperoxidase increases gradually at later times using PMA as the cell stimulator.
Thus, pholasin can be used to measure the formation of superoxide anion radicals as well as the release of myeloperoxidase in suspensions of stimulated phagocytosing cells containing a low number of cells.
Antibody techniques. Antibodies against myeloperoxidase can also be used to detect the enzyme release from stimulated PMNs [5]. The advantage of this approach is the high specificity. However, a sufficiently large number of cells (one million cells or more) have to be used. Furthermore, time-resolved measurements are hardly possible. Concomitant results concerning the release of myeloperoxidase from stimulated PMNs have been obtained using the pholasin luminescence and the detection of myeloperoxidase by specific antibodies [78].
Determination of MPO activity in cell supernatants. There are numerous methods to detect active MPO in solution. These methods are based on the determination either of its chlorinating [79] or oxidizing activity [80]. Although theses methods work usually well in enzyme solutions, many pitfalls have been observed in cell suspensions. There was a permanent loss of enzyme activity after degranulation of azurophilic granules. A progressive inactivation of myeloperoxidase and other enzymes released from azurophilic granules [81-84] or attachment to biological membranes [38, 49] has been discussed as potential reasons for these findings.
Thus, myeloperoxidase is a unique, multifunctional enzyme involved in both host defense and tissue damage at inflammatory sites. It produces not only oxidative equivalents, but contributes also to the regulation in general response to invading microorganisms.
I deeply acknowledge valuable discussions on myeloperoxidase topics with my colleagues and friends in Moscow, Vienna, Leipzig, and other places.
This work was supported by the German Research Foundation (Grant GL 199/4; Postgraduate Training Program “Mechanisms and Applications of Nonconventional Oxidation Reactions”).
REFERENCES
1.Fantone, J. C., and Ward, P. A. (1982) Am. J.
Pathol., 107, 397-418.
2.Allen, R. C., and Stevens, D. L. (1992) Curr.
Opin. Infect. Dis., 5, 389-398.
3.Edwards, S. W. (1994) Biochemistry and
Physiology of the Neutrophil, Cambridge University Press.
4.Borregaard, N., Miller, L. J., and Springer, T. A.
(1987) Science, 237, 1204-1206.
5.Sengeloev, H., Kjeldsen, L., and Borregaard, N.
(1993) J. Immunol., 150, 1535-1543.
6.Babior, B. M., Kipnes, R. S., and Curnutte, J. T.
(1973) J. Clin. Invest., 52, 741-744.
7.Segal, A. W., and Abo, A. (1993) Trends Biochem.
Sci., 18, 43-47.
8.Carlson, M. G. C., Peterson, C. G. B., and Venge,
P. (1985) J. Immunol., 134, 1875-1879.
9.Sakamaki, K., Tomonaga, M., Tsukui, K., and Nagata,
S. (1989) J. Biol. Chem., 264, 16828-16836.
10.Nauseef, W. M., and Malech, H. L. (1986)
Blood, 67, 1504-1507.
11.Andrews, P. C., Parnes, C., and Krinsky, N. I.
(1984) Arch. Biochem. Biophys., 228, 439-442.
12.Fiedler, T. J., Davey, C. A., and Fenna, R. E.
(2000) J. Biol. Chem., 275, 11964-11971.
13.Kettle, A. J., and Winterbourn, C. C. (1994)
J. Biol. Chem., 269, 17146-17151.
14.Furtmueller, P. G., Burner, U., Jantschko, W.,
Regelsberger, G., and Obinger, C. (2000) FEBS Lett., 484,
139-143.
15.Floris, R., and Wever, R. (1992) Eur. J.
Biochem., 207, 697-702.
16.Furtmueller, P. G., Burner, U., Jantschko, W.,
Regelsberger, G., and Obinger, C. (2000) Redox Report, 5,
173-178.
17.Dolphin, D., and Felton, R. H. (1974) Acc.
Chem. Res., 7, 26.
18.Van Dalen, C. J., Whitehouse, M. W., Winterbourn,
C. C., and Kettle, A. J. (1997) Biochem. J., 327,
487-492.
19.Marquez, L. A., and Dunford, H. B. (1995) J.
Biol. Chem., 270, 30434-30440.
20.Burner, U., Jantschko, W., and Obinger, C. (1999)
FEBS Lett., 443, 290-296.
21.Burner, U., Furtmueller, P. G., Kettle, A. J.,
Koppenol, W. H., and Obinger, C. (2000) J. Biol. Chem.,
275, 20597-20601.
22.Abu-Soud, H. M., and Hazen, S. L. (2000) J.
Biol. Chem., 275, 37524-37532.
23.Podrez, E. A., Abu-Soud, H. M., and Hazen, S. L.
(2000) Free Rad. Biol. Med., 28, 1717-1725.
24.Zgliczynski, J. M., Selvaraj, R. J., Paul, B. B.,
Stelmazynska, T., Poskitt, P. K. F., and Sbarra, A. J. (1977) Proc.
Soc. Exp. Biol. Med., 154, 418-422.
25.Bakkenist, A. R. J., De Boer, J. E. G., Plat, H.,
and Wever, R. (1980) Biochim. Biophys. Acta, 613,
337-348.
26.Held, A. M., Halko, D. J., and Hurst, J. K.
(1978) J. Am. Chem. Soc., 100, 5732-5740.
27.Candeias, L. P., Patel, K. B., Stratford, M. R.
L., and Wardman, P. (1993) FEBS Lett., 333, 151-153.
28.Ramos, C. L., Pou, S., Britigan, B. E., Cohen, M.
S., and Rosen, G. M. (1992) J. Biol. Chem., 267,
8307-8312.
29.Osipov, A. N., Yakutova, E. S., and Vladimirov,
J. A. (1993) Biofizika, 38, 383-388.
30.Wardman, P., and Candeias, L. P. (1996) Rad.
Res., 145, 523-531.
31.Eiserich, J. P., Hristova, M., Cross, C. E.,
Jones, A. D., Freeman, B. A., Halliwell, B., and van der Vliet, A.
(1998) Nature, 391, 393-397.
32.Arnhold, J., Furtmueller, P. G., Regelsberger,
G., and Obinger, C. (2001) Eur. J. Biochem., 268,
5142-5148.
33.Furtmueller, P. G., Arnhold, J., Jantschko, W.,
Pichler, H., and Obinger, C. (2003) Biochem. Biophys. Res.
Commun., 301, 551-557.
34.Winterbourn, C. C. (1985) Biochim. Biophys.
Acta, 840, 204-210.
35.Arnhold, J., Mueller, S., Arnold, K., and
Sonntag, K. (1993) J. Biolum. Chemilum., 8, 307-313.
36.Thomas, E. L., Jefferson, M. M., and Grisham, M.
B. (1982) Biochemistry, 21, 6299-6308.
37.Test, S. T., Lambert, M. B., Ossanna, P. J.,
Thoene, J. G., and Weiss, S. J. (1982) J. Clin. Invest.,
74, 1341-1349.
38.Johansson, M. W., Patarroyo, M., Oeberg, F.,
Siegbahn, A., and Nilsson, K. (1997) J. Cell Sci., 110,
1133-1139.
39.Arnhold, J., Osipov, A. N., Spalteholz, H.,
Panasenko, O. M., and Schiller, J. (2001) Free Rad. Biol. Med.,
9, 1111-1119.
40.Arnhold, J., Osipov, A. N., Spalteholz, H.,
Panasenko, O. M., and Schiller, J. (2002) Biochim. Biophys.
Acta, 1572, 91-100.
41.Panasenko, O. M., Spalteholz, H., Schiller, J.,
and Arnhold, J. (2003) Free Rad. Biol. Med., 34,
553-562.
42.Panasenko, O. M., Arnhold, J., Vladimirov, J. A.,
Arnold, K., and Sergienko, V. I. (1997) Free Rad. Res.,
27, 1-12.
43.Panasenko, O. M., and Arnhold, J. (1999) Free
Rad. Res., 30, 479-487.
44.Heinecke, J. W., Li, W., Francis, G. A., and
Goldstein, J. A. (1993) J. Clin. Invest., 91,
2866-2872.
45.Savenkova, M. L., Mueller, D. M., and Heinecke,
J. W. (1994) J. Biol. Chem., 269, 20394-20400.
46.Biasucci, L. M., D'Onofrio, G., Liuzzi, G., Zini,
G., Monaco, C., Caligiuri, G., Tommasi, M., Rebuzzi, A. G., and Maseri,
A. (1996) J. Am. Coll. Cardiol., 27, 611-616.
47.Deby-Dupont, G., Deby, C., and Lamy, M. (1999)
Intensivmedizin Notfallmedizin, 36, 500-513.
48.Daphna, E. M., Michaela, S., Eynat, P., Irit, A.,
and Rimon, S. (1998) Mol. Cell Biochem., 183, 55-61.
49.Baldus, S., Eiserich, J. P., Mani, A., Castrom,
L., Figueroa, M., Chumley, P., Ma, W., Tousson, A., White, R., Bullard,
D. C., Brennan, M.-L., Lusis, A. J., Moore, K. P., and Freeman, B. A.
(2001) J. Clin. Invest., 108, 1759-1770.
50.Cuzzocrea, S., Costantino, G., Mazzon, E., and
Caputi, A. P. (1999) Crit. Care Med., 27, 1524-1532.
51.Van der Vliet, A., Nguyen, M. N., Shigenaga, M.
K., Eiserich, J. P., Marelich, G. P., and Cross, C. E. (2000) Am. J.
Physiol. Lung Cell. Mol. Physiol., 279, L537-L546.
52.Baldus, S., Eiserich, J. P., Brennan, M.-L.,
Jackson, R. M., Alexander, C. B., and Freeman, B. A. (2002) Free
Rad. Biol. Med., 33, 1010-1019.
53.Eiserich, J. P., Baldus, S., Brennan, M.-L., Ma,
W., Zhang, C., Tousson, A., Castro, L., Lusis, A. J., Nauseef, W. M.,
White, C. R., and Freeman, B. A. (2002) Science, 296,
2391-2394.
54.Nauseef, W. M. (1988) Hematol. Oncol. Clin.
North Am., 2, 135-158.
55.Rosen, H., and Klebanoff, S. (1976) J. Clin.
Invest., 58, 50-60.
56.Stendahl, O., Coble, B.-I., Dahlgren, C., Hed,
J., and Molin, L. (1984) J. Clin. Invest., 73,
366-373.
57.Podrez, E. A., Schmitt, D., Hoff, H. F., and
Hazen, S. L. (1999) J. Clin. Invest., 103, 1547-1560.
58.Nagra, R., Becher, B., Tourtellotte, W. W.,
Antel, J. P., Gold, D., and Palidino, T. (1997) Neuroimmunology,
78, 97-107.
59.Reynolds, W. F., Rhees, J., Maciejewski, D.,
Paladino, T., Sieburg, H., Maki, R. A., and Masliah, E. (1999) Exp.
Neurol., 155, 31-41.
60.Jolivalt, C., Leininger-Muller, B., Drozdz, R.,
Naskalski, J. W., and Siest, G. (1996) Neurosci. Lett.,
210, 61-64.
61.Daugherty, A., Dunn, J. L., Rateri, D. L., and
Heinecke, J. W. (1994) J. Clin. Invest., 94, 437-444.
62.Malle, E., Waeg, G., Schreiber, R., Grone, E. F.,
Sattler, W., and Grone, H. J. (2000) Eur. J. Biochem.,
267, 4495-4503.
63.Nakajima, K., Powers, J. C., Ashe, B. M., and
Zimmermann, M. (1979) J. Biol. Chem., 254, 4027-4032.
64.Allen, R. C., Stjernholm, R. L., and Steele, R.
H. (1972) Biochem. Biophys. Res. Commun., 47,
679-684.
65.Baxendale, J. H. (1973) J. Chem. Soc. Faraday
Trans. I, 69, 1665-1677.
66.Eriksen, T. E., Lind, J., and Meroenyi, G. (1981)
J. Chem. Soc. Faraday Trans. I, 77, 2125-2135.
67.Meroenyi, G., Lind, J., and Eriksen, T. E. (1990)
J. Biolum. Chemilum., 5, 53-56.
68.Meroenyi, G., Lind, J., and Eriksen, T. E. (1986)
J. Am. Chem. Soc., 108, 7716-7726.
69.Dahlgren, C., and Stendahl, O. (1982) Infect.
Immun., 39, 34-39.
70.Dahlgren, C. (1989) Free Rad. Biol. Med.,
6, 399-403.
71.Radi, R., Cosgrove, T. P., Beckamn, J. S., and
Freeman, B. A. (1993) Biochem. J., 290, 51-57.
72.Fukuyama, N., Ichimori, K., Su, Z., Ishida, H.,
and Nakazawa, H. (1996) Biochem. Biophys. Res. Commun.,
224, 414-419.
73.Henry, J. P., and Michelson, A. M. (1973)
Biochimie, 55, 83-93.
74.Roberts, P. A., Knight, J., and Campbell, A. K.
(1987) Analyt. Biochem., 160, 139-148.
75.Dunstan, S. L., Sala-Newby, G. B., Fajardo, A.
B., Taylor, K. M., and Campbell, A. K. (2000) J. Biol. Chem.,
275, 9403-9409.
76.Reichl, S., Arnhold, J., Knight, J., Schiller,
J., and Arnold, K. (2000) Free Rad. Biol. Med., 28,
1555-1563.
77.Wittko-Sarsat, V., Nguyen, A. T., Knight, J., and
Descamps-Latscha, B. (1992) Free Rad. Biol. Med., 13,
83-88.
78.Reichl, S., Vocks, A., Petkovich, M., Schiller,
J., and Arnhold, J. (2001) Free Rad. Res., 35,
723-733.
79.Kettle, A. J., and Winterbourn, C. C. (1994)
Meth. Enzymol., 233, 502-512.
80.Klebanoff, S. J., Waltersdorph, A. M., and Rosen,
H. (1984) Meth. Enzymol., 105, 399-403.
81.Voetman, A. A., Weening, R. S., Hamers, M. N.,
Meerhof, L. J., Bot, A. M., and Roos, D. (1981) J. Clin.
Invest., 67, 1541-1549.
82.Edwards, S. W., Say, J. E., Taylor, J., and Hart,
C. A. (1988) J. Clin. Lab. Immunol., 27, 97-102.
83.Bach, M. K., Brashler, J. R., Sanders, M. E., and
Bienkowski, M. J. (1991) J. Immunol. Meth., 142,
243-250.
84.King, C. C., Jefferson, M. M., and Thomas, E. L.
(1997) J. Leukoc. Biol., 61, 293-302.