Analysis of Mitochondrial DNA: Taxonomic and Phylogenetic Relationships in Two Fish Taxa (Pisces: Mugilidae and Cyprinidae)
A. V. Semina1*, N. E. Polyakova1, and Vl. A. Brykov1,2
1Zhirmunsky Institute of Marine Biology, Far Eastern Branch of the Russian Academy of Sciences, ul. Pal'chevskogo 17, 690041 Vladivostok, Russia; fax: (4232) 310-900; E-mail: alicesem@rambler.ru; neonpol@rambler.ru; vlbrykov@mail.ru2Far Eastern State University, ul. Oktyabr'skaya 27, 690600 Vladivostok, Russia
* To whom correspondence should be addressed.
Received July 18, 2007
To solve some systematic questions as well as to study genetic variability and evolutionary relationships in two groups of fish belonging to the Mugilid (Mugilidae) and Cyprinid (Cyprinidae) families, we have used restriction fragment length polymorphism analysis of mitochondrial DNA (mtDNA) fragments amplified in polymerase chain reaction. The analysis of three mtDNA fragments of 7220 bp total length of six Mugilid species has shown that Mediterranean Liza aurata, L. ramada, L. saliens, and Chelon labrosus form a common cluster, L. aurata and C. labrosus being the closest relatives, whereas L. haematocheilus (syn. C. haematocheilus) of the Sea of Japan forms a sister group to the Mediterranean cluster. It was found that Chelon and Liza genera are paraphyletic, and therefore their division into two genera is unnatural and they should be synonymized. According to priority, Liza species should be ascribed to Chelon genus. Mugil cephalus is the most distant compared to the rest of the species studied. The level of genetic divergence between allopatric samples of M. cephalus from the Sea of Japan and the Mediterranean Sea has proved to be very high--4.5% of nucleotide substitutions. The analysis of four mtDNA fragments of 9340 bp total length of six Cyprinid species has shown that L. waleckii is the most genetically distant. Pseudaspius leptocephalus is a sister group to Tribolodon species. All Tribolodon species form a common cluster with T. sachalinensis as a root. The remaining species form two branches, one of which includes T. nakamurai and T. brandtii, another one combines T. hakonensis and a new form of Tribolodon revealed that is close to T. hakonensis by its mtDNA (2.4% of nucleotide substitutions). This new form might be an independent species.
KEY WORDS: Mugilidae, Cyprinidae, Tribolodon, PCR-RFLP, mtDNA, phylogenyDOI: 10.1134/S0006297907120085
Abbreviations: mtDNA) mitochondrial DNA; PCR) polymerase chain reaction; RFLP) restriction fragment length polymorphism.
Phylogenetic studies as a basis for systematic reconstructions and
analyses of direction and rate of evolution take an important place in
biological sciences [1]. Great interest in
phylogenetic reconstructions and problems connected to them gave rise
to various divisions, tendencies, and methods that are used to clarify
and specify the similarity and differences among taxa in all levels of
organization. The fundamental thesis of the evolutionary hypothesis
states that changes on molecular level of organisms lead to changes on
other levels. In a classic work of Spirin, Belozersky, et al. 50 years
ago it was shown that “... DNA has pronounced species specificity,
close species having fewer differences in DNA composition compared to
systematically distant ones” [2]. The
development of this inference has resulted in wide use of comparative
analysis of nucleotide sequences to solve taxonomic and systematic
problems in all groups of the organic world [3].
The reason for usage of this approach is the fact that in a process of
divergence, DNA accumulates nucleotide substitutions (mutations) that
lead to biochemical, physiological, morphological, and ethological
changes and, eventually, to the origin of new species and groups of
species.
To compare DNA sequences and uncover differences various methods are used at the present time, ranging from different variants of DNA-DNA-hybridization to direct comparison of nucleotide sequences of genes and genome regions [3]. The accumulated empirical data allow the assertion that molecular-genetic analysis can be successfully used to solve many disputable questions of systematics and phylogeny of taxa. Molecular methods also reveal cryptic species indistinguishable by morphological characters.
One of the most widely used approaches nowadays is comparative analysis of mitochondrial DNA (mtDNA). The mtDNA is characterized by a number of traits that make it suitable for these purposes [4]. They include the small size of the molecule [5]; the high rate of evolution of mitochondrial gene sequences, which is 5-10-fold faster than in nuclear loci [6]; haploid maternal inheritance; and lack of recombination [7]. Analysis of mutations in mtDNA and their distribution in the species habitat allows not only the differentiation of the taxa but also the retrospective reconstruction of the consecution of the origin of taxa and intra-species groups as well as the estimation of the approximate time of their divergence [4].
In the present study, we have used RFLP (restriction fragment length polymorphism)-analysis of mtDNA fragments amplified in polymerase chain reaction (PCR). The advantage of this method is the possibility to analyze extended mtDNA fragments that have substantially different rates of evolution and, correspondingly, different levels of resolution for phylogenetic reconstructions [8]. This approach has been used to analyze species in two fish taxa whose taxonomic status and phylogenetic relationships have not been unambiguously established: gray mullets (family Mugilidae) and Far Eastern redfins of Tribolodon genus (family Cyprinidae).
Gray mullets are distributed worldwide and inhabit marine, estuarine, and freshwater environments in all latitudes except the polar regions. The family Mugilidae (Pisces, Mugiliformes) includes 14 genera and 64 valid species. Most of them are representatives of Liza and Mugil genera. There are also 17 nominal taxa whose taxonomic status is uncertain [9]. The precise phylogenetic relationships of some species and genera in this family remain enigmatic because very few morphological characters are suitable to unambiguously establish the relationships among them due to their conservative morphology [10]. Moreover, there are substantial disagreements in Mugilid systematics [9, 11, 12].
Far Eastern redfins of Tribolodon genus (Pisces: Cypriniformes) are endemic for the Far Eastern Seas. Tribolodon species are the only group of Cyprinids that has anadromous ecotypes adapted to fattening at the ocean salinity and forming stable freshwater resident ecotypes [13]. At present four species of Far Eastern redfins are described. Three of them, Tribolodon hakonensis, T. brandtii, and T. sachalinensis, have been known for a long time and are common in the ichthyofauna of the Sea of Japan, south of the Okhotsk Sea, and the east coast of the Japanese Islands. The fourth species, T. nakamurai, which is endemic to some rivers of central Honshu Island, was described only in 2000 [14]. The study of genetic diversity and phylogenetic relationships of Tribolodon species has been previously conducted using allozyme methods [15-18]. However, many questions of Cyprinid evolutionary history and taxonomy still remain unsolved.
Thus, the problems existing in phylogeny and systematics of these two fish groups have given occasion to the present research.
MATERIALS AND METHODS
In the present study we have analyzed mtDNA of six Mugilid species: Mugil cephalus Linnaeus, 1758 and Liza haematocheilus (syn. Chelon haematocheilus) Temminck et Schlegel, 1845 from the Sea of Japan, as well as M. cephalus, L. aurata Risso, 1810, L. ramada Risso, 1826, L. saliens Risso, 1810, and Chelon labrosus Risso, 1826 from the Mediterranean Basin.
Among Cyprinids, mtDNA of four species of Far Eastern redfins have been compared: south and north forms of T. hakonensis Gunther, 1880 [19], T. brandtii Dybowski, 1872, T. sachalinensis Nikolsky, 1889, and T. nakamurai Doi et Shinzawa, 2002, as well as close taxa used as outgroups: Leuciscus waleckii Dybowsky, 1869 and Pseudaspius leptocephalus Pallas, 1776.
The information about the sample collection sites is presented in Tables 1 and 2. Total DNA was extracted from the tissue pieces fixed in 95% ethanol according to the standard protocol [20]. The variability of mtDNA was revealed by PCR-RFLPs. Primers designed by Gharrett et al. [21] were used to amplify different mtDNA fragments. PCR was performed in 50 µl reactions with 2 units of Taq polymerase, 5 µl of 10× Taq buffer (Sibenzyme, Russia), 2 mM of each dNTP, 0.25 µM of each primer, and approximately 150 ng of genomic DNA as a template. Amplifications began with preliminary denaturation at 94°C (5 min), followed by strand denaturation at 94°C (0.5 min), primer annealing at 50°C (1 min), and primer extension at 72°C (2.4 min) repeated for 35 cycles and final extension at 68°C (4 min).
Table 1. Geographic localization and sample
size of Mugilid samples collected in 2002-2004

Table 2. Geographic localization and sample
size of Cyprinid samples collected in 2000-2005

For the Mugilid analysis, three mtDNA fragments were amplified, two of them coding five nicotinamide dinucleotide dehydrogenase subunits (ND3/ND4L/ND4 and ND5/ND6), the other coding 12S and 16S ribosomal RNA (12S/16S rRNA). PCR-products were digested by the set of 13 restriction endonucleases: AvaII, BsuRI, Cfr13I, DdeI, Hin6I, HinfI, MboI, MspI, MvaI, RsaI, StyI, TaqI, and VspI (Fermentas, Lithuania; Sibenzyme). For the Cyprinid analysis, four mtDNA fragments were amplified, coding ND3/ND4L/ND4, ND5/ND6, 12S/16S rRNA, as well as two subunits of ATPase and cytochrome oxidase III (ATP6/ATP8/COIII). Each of them was analyzed by 13 restriction endonucleases: Hin6I, MspI, VspI, PstI, AvaI, Cfr13I, MboI, HindIII, RsaI, BsuRI, MvaI, HinfI, and StyI (Fermentas; Sibenzyme).
Digested fragments were separated electrophoretically through 2% agarose gels (one part of regular agarose (Sigma, USA) and two parts of SynergelTM (Diversified Biotech, Inc., USA)) in 0.5× Tris-borate buffer [18], stained with ethidium bromide, and visualized under UV light. As molecular markers of length, we used lambda phage DNA hydrolyzed by PstI and 100 bp ladder standard included in each gel.
Each restriction pattern obtained by each restriction enzyme was designated by a letter. Composite haplotypes of each individual on each fragment were summarized by a letter-code, 39 for the Mugilids and 50 for the Cyprinids (not 52 since HindIII and PstI have not revealed restriction sites in 12S/16S rRNA mtDNA fragment of Cyprinids). The data obtained from restriction fragments were converted into restriction site binary matrixes (gain or loss of the site as a result of mutation). Phylogenetic analysis was performed in PAUP* 4.0b10 (tree bisection-reconnection method of branch-swapping with 50 random-addition sequences and 1000 bootstrap replicates) [22]. The haplotype and nucleotide diversity as well as the level of nucleotide divergence within and among the species were calculated using the REAP program package [23].
RESULTS
Family Mugilidae. The analysis of Mugilid mtDNA has revealed 434 restriction sites over a 7220 bp segment. This segment comprises on average 45% of the whole fish mitochondrial genome [8]. Of 434, 368 sites proved to be parsimony informative. The approximate size of 12S/16S rRNA fragment of M. cephalus (2430 bp) differs from those of Liza and Chelon representatives (2400 bp) by about 30 bp. Among 88 individuals of six Mugilid species studied, 31 species-specific composite haplotypes have been detected: L. haematocheilus having seven, L. aurata - 12, C. labrosus - four, M. cephalus - six, L. ramada - one, and L. saliens - one haplotype.
The level of pair-wise mtDNA sequence divergence of the six species studied varies from 26.9-28.9% (between M. cephalus and L. haematocheilus) to 8.4% (between L. aurata and C. labrosus) of nucleotide substitutions (Table 3). Mugil cephalus is equidistant from all the Liza-Chelon species - from 26.1 to 28.9% of nucleotide substitutions. Differences among the species mtDNA within the Liza-Chelon cluster comprise 8.4-14.6%. Mugil cephalus haplotypes of the Sea of Japan and the Mediterranean Sea form two branches of the same cluster that precisely align with the geographical source of these populations (Fig. 1). Mugil cephalus of the Sea of Japan and Mediterranean had no common composite haplotypes. Five out of 13, 10 out of 13, and 11 out of 13 restriction endonucleases revealed differences between the two samples of M. cephalus in 12S/16S rRNA, ND3/ND4L/ND4, and ND5/ND6 mtDNA fragments, respectively.
Table 3. Nucleotide divergence in Mugilid
samples
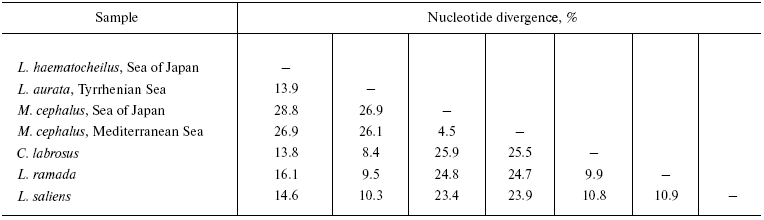
A consensus tree (L = 496, CI = 0.77, RI = 0.57) with the branch support values is shown in Fig. 1. It displays three main lineages: (1) L. haematocheilus branch; (2) a group including L. aurata, L. ramada, L. saliens, and C. labrosus; and (3) M. cephalus branch. Mugil cephalus is the most genetically distant species, while L. haematocheilus and Mediterranean Liza and Chelon species are sister groups.Fig. 1. Consensus tree (L = 496, CI = 0.77, RI = 0.57) illustrating Mugilid phylogenetic relationships with 1000 replicate bootstrap support.
Family Cyprinidae. The analysis of Cyprinid mtDNA has revealed 455 restriction sites over a 9340 bp segment. This segment comprises on average 58% of the whole fish mitochondrial genome [8]. Of 455, 335 sites proved to be parsimony informative. Among 109 individuals of six species studied in the present work, 53 species-specific composite haplotypes were scored: T. sachalinensis having seven, T. brandtii - 13, south form of T. hakonensis - four, north form of T. hakonensis from Sakhalin Island - 12, north form of T. hakonensis from Khabarovsky Region - 11, L. waleckii - four, T. nakamurai - one, and P. leptocephalus - one haplotype.
The level of pair-wise mtDNA sequence divergence among the six species studied varies from 16.3% (L. waleckii vs. T. hakonensis and T. nakamurai) to 5.2% (between T. nakamurai and T. brandtii) of nucleotide substitutions (Table 4). The mtDNA of L. waleckii differs from all the Tribolodon species by about the same number of nucleotide substitutions - from 14.9 to 16.8%, mtDNA of P. leptocephalus - from 7.7 to 10.6%.
Table 4. Nucleotide divergence in Cyprinid
samples

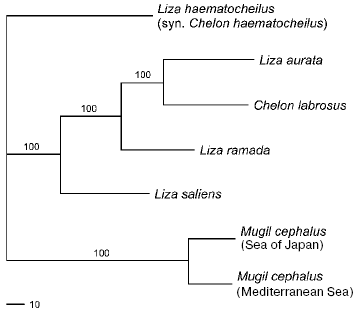
A consensus tree (L = 478, CI = 0.76, RI = 0.49) with branch support values is presented in Fig. 2. It forms three main clusters: (1) L. waleckii branch; (2) P. leptocephalus branch; and (3) a group including T. sachalinensis, T. nakamurai, T. brandtii, as well as the south and north forms of T. hakonensis. Among the species studied, L. waleckii is the most genetically distant species. Genetic differences between the south (Primorye) and north (Sakhalin and Khabarovsky Region) samples of T. hakonensis appeared to be high - 2.4% of nucleotide substitutions (Table 4). Nucleotide diversity of the north form is 0.013146 (Sakhalin Island) and 0.010139 (Khabarovsky Region), nucleotide diversity of the south form is 0.001130.
Fig. 2. Consensus tree (L = 478, CI = 0.76, RI = 0.49) illustrating Cyprinid phylogenetic relationships with 1000 replicate bootstrap support.
DISCUSSION
Results obtained on the basis of comparative mtDNA analysis have allowed the clarification of some controversial questions of systematics and phylogeny of the two fish taxa studied and the acquisition of interesting new data.
Family Mugilidae. The present genetic analysis has revealed a high level of mtDNA differentiation of all the species studied. On the whole, the results are similar to the schemes based on comparisons of hemoglobins [24], allozyme analysis [25-27], PCR-RFLP-analysis of mtDNA [28, 29], comparison of 16S rRNA [27], karyologic analysis [30], and comparison of nucleotide sequences [31]. However, there are some differences.
The highest level of divergence was observed between M. cephalus and the other species studied, which confirms the current conception on Mugilid systematic relationships [9]. All the Mediterranean Liza species have about the same genetic distances among each other (8-10% of nucleotide substitutions). Liza haematocheilus and Mediterranean species are sister taxa (Fig. 1). At the same time, the number of its nucleotide substitutions compared to most of the species is about one and a half times higher (14-16%). This result may seem to prove the assumption that L. haematocheilus belongs to the different genus - Chelon [9]. However, the comparison of mtDNA of L. haematocheilus and C. labrosus, the representative of this genus, shows the same amount of difference - about 14%, while mtDNA of C. labrosus and Mediterranean species of Liza genus differs by 8-11% (Table 3). Our data testifies to the paraphyletic origin of L. haematocheilus and C. labrosus mtDNA and, thus, to the absence of reasons to unify them into the same genus.
The results evidence Chelon and Liza representatives' close genetic relation. Many other researchers also state the closeness of these genera. Comparative studies of the pharyngobranchial organ [32] and cytogenetic data [30, 33] have not found any substantial differences between C. labrosus and the three Mediterranean Liza species. In the studies based on genetic analysis of Mediterranean mullets, C. labrosus clusters either with L. saliens [28, 31], or L. aurata [27], or joins the L. ramada-L. aurata cluster [34]. Therefore, taking into account the scientific issue data, our results point out the necessity for taxonomic revision and synonymization of Chelon and Liza genera. According to the priority, all these species should be ascribed to Chelon genus [11].
The values of divergence (Table 3) obtained in the present work are higher than in the studies of Caldara et al. [31] and Papasotiropoulos et al. [28], which might be explained by the fact that both groups of authors used short (400-630 bp) and conservative (COI, cytochrome b, and 12S and 16S rRNA) mtDNA fragments. It is supported by the fact that if our data are computed basing only on highly conservative 12S/16S rRNA region, the level of divergence abruptly decreases to 19.3-18.6% between M. cephalus and L. haematocheilus, to 1.8% between L. aurata and C. labrosus, and to 1.6% between C. labrosus and L. ramada. This supports the idea that the information based exclusively on small and conservative DNA fragments is not enough to reconstruct phylogenetic relationships among closely related organisms.
One more interesting inference follows from the comparison of allopatric populations of M. cephalus mtDNA from the Mediterranean and the Sea of Japan. The level of their differentiation has proved to be high, i.e. 4.5% of nucleotide substitutions. Such a deep level of divergence and the absence of common haplotypes, along with the fact that 26 out of 39 restriction enzymes used differentiate both M. cephalus samples, testify to the long genetic isolation of these populations. In other researchers' studies, it was shown that despite the cosmopolite distribution and morphological identity, M. cephalus has a pronounced population genetic structure [25, 35-37]. However, since the genetic subdivision of M. cephalus populations contradicts its great morphological uniformity of this species on its habitat [25], the question about the systematic significance of the divergence level observed and conspecificity of these populations demand further investigations.
Family Cyprinidae. In general, the consecution of divergence events of Tribolodon species and the amount of their genetic divergence level are similar to the schemes based on allozyme and rRNA sequence data [15-18].
The most genetically distant species is L. waleckii. Its divergence from the rest of the species studied lies within the bounds of 14.8 and 17.2%. The close relationship of P. leptocephalus and Tribolodon species (7.8-11.1%) confirms the data of Kartavtsev et al. [16], Sakai et al. [17, 18], and Sasaki et al. [38]. Differences among the mtDNA of the Tribolodon species are about the same: 10.9% between T. hakonensis vs. two other species--T. brandii and T. sachalinensis, 8.5% between T. brandii and T. sachalinensis.
In comparing the north and south forms of T. hakonensis, deep genetic differences--about 2.5% of mtDNA nucleotide substitutions--have been shown. The number of north form composite haplotypes (12 and 11) is three times higher than the south form's (four), all of them being strictly form specific. The level of nucleotide diversity of the north form (0.013146 and 0.010139) is an order of magnitude higher than that of the south form (0.001130) taking into account that the number of the specimens analyzed was equal in each sample. These facts prove the existence of long genetic isolation between the forms during many generations. Additional evidence follows from the study of Sakai et al. [17], who showed that T. hakonensis populations from Sakhalin, Khabarovsky Region, and Japan on one hand, and Korea and Primorye on the other, have fixed differences by Prot-2 locus, while T. brandii, whose species status is undoubted, had no diagnostic loci. These authors showed that in the narrow Korean Strait between the southern Korean Peninsula and the Japanese Archipelago both alleles occurred in approximately the same proportion, which is evidence of the two forms' sympatric habitat. Thus, the absence of genetic drift under the probable sympatry enables the assumption that these two forms are distinct species.
Therefore, this group of fishes also needs a taxonomic revision in the light of molecular genetic data reconsidering the status of the north and south forms of T. hakonensis and perhaps of Pseudaspius and Tribolodon genera.
The authors thank A. R. Rossi, H. Sakai, N. S. Romanov, S. M. Dolganov, and Y. F. Kartavtsev for their help in sample collection.
This study was partly supported by FEB RAS grants (06-III-V-06-212 and 06-P10-015) and Russian Science Support Foundation.
REFERENCES
1.Mayr, E. (1963) Animal Species and
Evolution, Harvard University Press, Cambridge MA, USA.
2.Spirin, A. S., Belozersky, A. N., Shugaeva, N. V.,
and Vanushin, B. F. (1957) Biokhimiya, 22, 744-753.
3.Hills, D. M., Moritz, C., and Mable, B. K. (eds.)
(1996) Molecular Systematics, Sinauer Associates, Inc.,
Sunderland, MA, USA.
4.Avise, J. C. (2000) Phylogeography. The History
and Formation of Species, Harvard University Press, Cambridge.
5.Attardi, G. (1985) Int. Rev. Cytol.,
93, 93-145.
6.Brown, W. M., George, M., Jr., and Wilson, A. C.
(1979) Proc. Natl. Acad. Sci. USA, 76, 1967-1971.
7.Birky, C. W. (2001) Annu. Rev. Genet.,
35, 125-148.
8.Churikov, D., Matsuoka, M., Luan, X., Gray, A. K.,
Brykov, Vl. A., and Gharrett, A. J. (2001) Mol. Ecol.,
10, 2329-2339.
9.Thomson, J. M. (1997) Mem. Queensl. Mus.,
41, 457-562.
10.Stiassny, M. (1993) Bull. Marine Sci.,
52, 197-219.
11.Schultz, L. P. (1946) Proc. U. S. Natl.
Mus., 96, 377-395.
12.Parin, N. V. (2003) Vopr. Ikhtiol.,
43, 418-419.
13.Gavrenkov, Y. I. (1987) Biology of Far Eastern
Redfins of Tribolodon Genus as a Perspective Object of the South
Primorye Aquaculture [in Russian]: Candidate's disseration,
VNIIPRKh, Moscow.
14.Doi, A., and Shinzawa, H. (2000) Ruffles Bull.
Zool., 48, 241-247.
15.Hanzawa, N., and Taniguchi, N. (1982) Fish.
Genet. Breed. Sci., 7, 26-30.
16.Kartavtsev, Y. F., Sviridov, V. V., Hanzawa, H.,
and Sazaki, T. (2002) Genetika, 38, 1285-1297.
17.Sakai, H., Goto, A., and Jeon, S.-R. (2002)
Zool. Sci., 19, 1291-1303.
18.Sakai, H., Ito, Y., Shedko, S. V., Safronov, S.
N., Frolov, S. V., Chereshnev, I. A., Jeon, S.-R., and Goto, A. (2006)
Zool. Sci., 23, 323-331.
19.Semina, A. V., Polyakova, N. E., and Brykov, Vl.
A. (2006) Dokl. Akad. Nauk, 407, 571-573.
20.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning: A Laboratory Manual, Cold Spring
Harbor Laboratory Press, N. Y.
21.Gharrett, A. J., Gray, A. K., and Brykov, V. A.
(2001) Fish. Bull., 99, 528-544.
22.Swofford, D. L. (2002) PAUP*. Phylogenetic
Analysis Using Parsimony (*and Other Methods). Version 4, Sinauer
Associates, Sunderland, Massachusetts.
23.McElroy, D., Moran, P., Birmingham, E., and
Kornfield, I. (1992) J. Heredity, 83, 157-158.
24.Rizotti, M. (1993) Trends Comp. Biochem.
Physiol., 1, 385-392.
25.Rossi, A. R., Capula, M., Crosetti, D., Sola, L.,
and Campton, D. E. (1998) Marine Biol., 131, 203-212.
26.Papasotiropoulos, V., Klossa-Kilia, E., Kilias,
G., and Alahiotis, S. (2001) Biochem. Genet., 39,
155-168.
27.Rossi, A. R., Ungaro, A., Innocentis, S. D.,
Crosetti, D., and Sola, L. (2004) Biochem. Genet., 42,
301-315.
28.Papasotiropoulos, V., Klossa-Kilia, E., Kilias,
G., and Alahiotis, S. (2002) Biochem. Genet., 40,
71-86.
29.Semina, A. V., Polyakova, N. E., Makhotkin, M.
A., and Brykov, Vl. A. (2007) Biol. Morya, 33,
223-228.
30.Cataduella, S., Civitelli, M. V., and Capanna, E.
(1974) Caryologia, 27, 93-105.
31.Caldara, F., Bargelloni, L., Ostellari, L.,
Penzo, E., Colombo, L., and Patarnello, T. (1996) Mol. Phylogenet.
Evol., 6, 416-424.
32.Harrison, I. J., and Howes, G. J. (1991) Bull.
Br. Mus. Nat. Hist. (Zool.), 57, 111-132.
33.Gornung, E., Cordisco, C. A., Rossi, A. R.,
Innocentiis, S. D., Crosetti, D., and Sola, L. (2001) Marine
Biol., 139, 55-60.
34.Murgia, R., Tola, G., Archer, S. N., Vallerga,
S., and Hirano, J. (2002) Marine Biotechnol., 4,
119-126.
35.Rossi, A. R., Capula, M., Crosetti, D., Campton,
D. E., and Sola, L. (1998) Marine Biol., 131,
213-218.
36.Crosetti, D., Nelson, W. S., and Avise, J. C.
(1994) J. Fish Biol., 44, 47-58.
37.Rocha-Olivares, A., Garber, N. M., and Stuck, K.
C. (2000) J. Fish Biol., 57, 1134-1149.
38.Sasaki, T., Kartavtsev, Y. P., Chiba, S. N.,
Uematsu, T., Sviridov, V. V., and Hanzawa, N. (2007) Genes Genet.
Syst., 82, 329-340.