Charge Separation in Rhodobacter sphaeroides Mutant Reaction Centers with Increased Midpoint Potential of the Primary Electron Donor
A. Yu. Khmelnitskiy*, R. A. Khatypov, A. M. Khristin, M. M. Leonova, L. G. Vasilieva, and V. A. Shuvalov
Institute of Basic Biological Problems, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; fax: (496) 773-0532; E-mail: hmelan@gmail.com* To whom correspondence should be addressed.
Received August 8, 2012; Revision received September 4, 2012
Primary charge separation dynamics in four mutant reaction centers (RCs) of the purple bacterium Rhodobacter sphaeroides with increased midpoint potential of the primary electron donor P (M160LH, L131LH, M197FH, and M160LH + L131LH + M197FH) have been studied by femtosecond transient absorption spectroscopy at room temperature. The decay of the excited singlet state in the wild-type and mutant RCs is complex and has two main exponential components, which indicates heterogeneity of electron transfer rates or the presence of reverse electron transfer reactions. The radical anion band of monomeric bacteriochlorophyll BA at 1020 nm was first observed in transient absorbance difference spectra of single mutants. This band remains visible, although with somewhat reduced amplitude, even at delays up to tens of picoseconds when stimulated emission is absent and the reaction centers are in the P+HA– state. The presence of this band in this time period indicates the existence of thermodynamic equilibrium between the P+BA–HA and P+BAHA– states. The data give grounds for assuming that the value of the energy difference between the states P*, P+BA–HA, and P+BAHA– at early times is of the same order of magnitude as the energy kT at room temperature. Besides, monomeric bacteriochlorophyll BA is found to be an immediate electron acceptor in the single mutant RCs, where electron transfer is hampered due to increased energy of the P+BA– state with respect to P*.
KEY WORDS: photosynthesis, charge separation, reaction center, femtosecond spectroscopy, electron transferDOI: 10.1134/S0006297913010070
Abbreviations: ΔA, absorption change (light-minus-dark); BA and BB, monomeric BChl in A- and B-chain, respectively; BChl, bacteriochlorophyll; BPheo, bacteriopheophytin; HA and HB, BPheo in A- and B-chain, respectively; P, primary electron donor, BChl dimer; PA and PB, BChl molecules within P; QA and QB, primary and secondary quinone, respectively; Rba., Rhodobacter; RC, reaction center.
The reaction center (RC) of purple bacteria is responsible for light
energy conversion into transmembrane electrical potential. Currently,
the high-resolution three-dimensional structure of the RC is resolved
for two species of purple bacteria – Blastochloris
(Rhodopseudomonas) (Blc. or Rps.) viridis
and Rhodobacter (Rba.) sphaeroides (Protein
Data Bank, files 2WJN [1] and 2J8C [2], respectively). The RC of Rba. sphaeroides
consists of three protein subunits (L, M, and H) and a number of
cofactors, which form two spatially symmetrical branches (A and B). The
primary electron donor, dimer P, consisting of two excitonically
coupled bacteriochlorophyll (BChl) molecules PA and
PB, is common to both branches. Each of the branches also
has monomeric BChl (BA or BB), bacteriopheophytin
(BPheo) (HA or HB), and quinone (QA or
QB). A non-heme iron atom and a carotenoid molecule are also
present as part of the RC. Only branch A is photochemically active in
the RC of purple bacteria. Electron transfer begins with charge
separation between the two molecules of excited dimer P, which leads to
formation of the
P*(PAδ+PBδ–)
state [3, 4]. Then, the
electron is transferred to the BChl BA to form the state
P+BA– and on to the molecule
HA, forming the state P+HA–
[5-9].
Participation of the monomer BChl BA as an intermediate acceptor in the primary charge separation logically follows from the position of BA between P and HA according to X-ray data [10, 11]. But for a long time experimental data providing direct evidence of the participation of BA in charge separation were not available. Largely because of this, an idea arose of possible virtual participation of a vacant electronic level of BA, located above the level of P* in the electron transfer from P to HA via a superexchange mechanism [12]. The difficulty in detecting the state P+BA– is due to the fact that the time of its formation is several times slower than the time of its depletion [13, 14]. This leads to a small population of the P+BA– state during the whole time of its existence, which does not exceed a few picoseconds. Another difficulty stems from the fact that, in the visible spectral range, where the vast majority of measurements have been carried out, the spectrum of P+BA– is strongly masked by spectra of other states including P*. Initially conclusive evidence for the existence the P+BA– state were obtained only in chemically modified RCs in which blocking of electron transfer to HA resulted in the accumulation of the P+BA– state in the picosecond time range [15]. Later an absorption band of the anion radical bacteriochlorophyll BA– at 1020 nm was detected in native reaction centers [16].
Experiments using mutagenesis have shown that the hydrogen bonds between histidine residues and carbonyl groups of BChl molecules comprising the special pair P significantly affect the redox potential of the primary donor, which in the wild-type RC is ~500 mV [17, 18]. Replacement of the phenylalanine at position M197 in the protein subunit by histidine leads to hydrogen bond formation between the histidine residue and the 2-acetyl group of PB and, as a result, the potential of the primary donor increases by ~125 mV [19]. Replacement of leucine by histidine at positions L131 and M160 leads to the formation of hydrogen bonds between histidine residues and 9-keto-groups of PA and PB, respectively, which results in an increase in the redox potential of P by ~80 mV in the first case and by ~60 mV in the second case [18, 20]. It was shown that the redox potential changes due to the amino acid substitutions are additive. Thus, the midpoint potential of the primary electron donor P in the mutant RC M160LH + L131LH + M197FH, carrying all three of these substitutions, increases by ~260 mV compared to the wild type.
The aim of this study was to investigate the participation of BChl BA molecules in the primary act of charge separation in the four mutant RCs of the purple bacterium Rba. sphaeroides with increased redox potential of the primary electron donor P by femtosecond spectroscopy.
MATERIALS AND METHODS
A genetic system for site-directed mutagenesis consisting of Rba. sphaeroides DD13 strain [21] deficient in RC and antennas systems synthesis and pRKP plasmids described elsewhere [22] was used. Mutagenesis was performed using the polymerase chain reaction method of overlapping DNA fragments as described previously [23]. Recombinant strains containing photosynthetic membranes with RCs and light-harvesting core antennas were grown semi-aerobically in Hattner media [24] in the dark. Tetracycline (1 µg/ml), kanamycin (5 µg/ml), and streptomycin (5 µg/ml) were added to the media.
The RCs of wild type and mutants were isolated from the corresponding recombinant strains of Rba. sphaeroides according to the procedure described previously [25]. The RCs were resuspended in buffer containing 20 mM Tris-HCl (pH 8.0) and 0.1% Triton X-100 and concentrated to an optical density of 0.5 at 870 nm (optical path length of 1 mm). Then sodium dithionite was added to a concentration of 5 mM, and the sample was illuminated with diffuse white light for 5 min to reduce primary quinone acceptor QA.
Femtosecond differential absorption spectra were obtained with a pump–probe spectrometer. A Ti:sapphire laser Tsunami (Spectra-Physics, USA) was used to obtain pulses with duration of ~35 fs at wavelength 800 nm and repetition rate 80 MHz. These pulses were used to seed an optical regenerative amplifier Spitfire (Spectra-Physics) to produce 40-fs pulses with energy of 0.9 mJ and a repetition rate of 50 Hz. The amplifier output beam was divided into two beams. The first beam was attenuated to energy of ~60 µJ and focused in a 5-mm thick cell containing water to generate a white light continuum. A small portion of the continuum (~4%) was used as a probe beam and a reference beam. The pump pulse with energy of ~0.54 mJ was focused on the 5-mm cell and the resulting continuum was used to excite the sample. All three pulses passed through a rotating quartz sample cell (thickness of 1 mm) with the pump and probe pulses overlapped. The time delay between the pump and probe pulses was varied by a computer-controlled optical delay PI-M531.DD (Physik Instrumente, Germany). On leaving the sample cell, the probe and reference pulses were directed to the entrance slit of a spectrograph Spectra Pro 2300i (Acton Research Corporation, USA). The spectra were recorded using an infrared camera Pixis 400BR CCD (Princeton Instruments, USA). Wavelengths shorter than 850 nm in the pump pulse were cut off with light filter RG850 (Newport, USA). The relative polarization of the pump and probe pulses was parallel. About 10-25% of the reaction centers were excited in the sample. Absorption difference spectra were obtained by averaging 1000 measurements at each time delay.
RESULTS
Figures 1-5 show the absorption difference spectra of RCs: the wild-type (Fig. 1) and mutants M160LH (Fig. 2), L131LH (Fig. 3), M197FH (Fig. 4), and M160LH + L131LH + M197FH (Fig. 5) in the region 920-1150 nm measured at different time delays after excitation with 40-fs pulses at wavelength 870 nm at room temperature. At time delay 20-60 fs, the absorption difference spectrum of the mutant and wild-type RCs reflects the absorption of a photon by the primary electron donor P (Figs. 1-5, dotted line). During this process, the primary electron donor P is promoted to excited state P* with stimulated emission shown in the spectrum as a negative absorption in the region <930 nm [26]. After about 60-80 fs, a shift of the stimulated emission to longer wavelength is observed, followed by the appearance of a positive absorption at 1100 nm, which reaches a maximum at 300-400 fs (Figs. 1-5, dot-dashed line). Recently, it was shown that these changes in the absorption spectra reflect the charge separation within the dimer of the primary electron donor P to form the P* (PAδ+PBδ–) state [3, 4], and the absorption at 1100 nm may belong to the PAδ+ cation [4].
With further increase in the time delay, a band at 1020 nm begins to appear in the absorption spectra of RCs of wild-type and single mutants, which is attributed to the absorption of the anion of the monomer bacteriochlorophyll of the active branch BA– (Figs. 1-4, dashed line) [15]. However, no significant decrease in the absorption at 1100 nm is observed, apparently due to the simultaneous formation of the P+ state, which shows broad and structureless absorption in this region [27]. Variance in the rate of development of the band at 1020 nm in the mutants reflects the rate of electron transfer from P* to BA and is consistent with the lifetime of the stimulated emission from state P* (Figs. 1-4, inset). At later delays, when stimulated emission from P* state is already absent, photoinduced changes in the absorption difference spectrum at 920-1150 nm for wild type (Fig. 1, solid line) and single mutants RCs (Figs. 2-4, solid line) are mainly associated with the absorption of the P+ and HA– states at 950 nm and the absorption of the residual BA– state at 1020 nm. Kinetic curves of the absorbance at 935 nm were satisfactorily approximated by two exponential functions with time constants of 2.9 ps (80%) and 15.5 ps (20%) for the wild-type RCs, 3.7 ps (70%) and 17 ps (30%) for the RCs of mutant M160LH, 4.5 ps (40%) and 20 ps (60%) for the RCs of mutant L131LH, 2.2 ps (50%) and 10.2 ps (50%) for M197FH RCs, and 12 ps (17%) and 137 ps (83%) for the triple mutant RCs M160LH + L131LH + M197FH (Figs. 1-5, inset).
Fig. 1. Absorbance difference spectra of wild-type RCs measured at different delay times. The inset shows photoinduced absorbance changes at 935 nm. The smooth, solid line is the result of fitting the data to a sum of two exponentials. Time constants and amplitudes of exponentials are also shown.
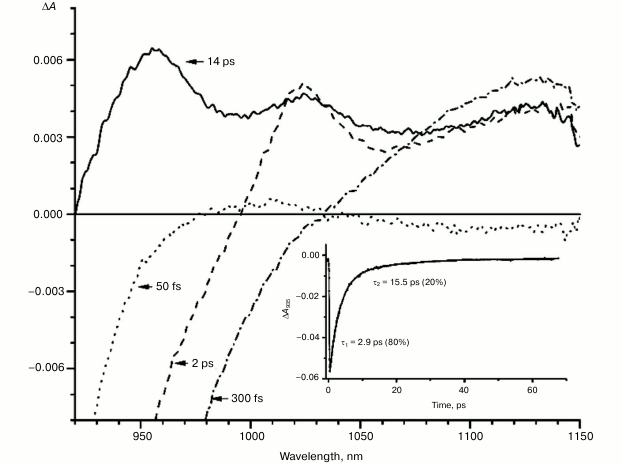
Fig. 2. Absorbance difference spectra of M160LH mutant RCs measured at different delay times. The inset shows photoinduced absorbance changes at 935 nm. The smooth, solid line is the result of fitting the data to a sum of two exponentials. Time constants and amplitudes of exponentials are also shown.
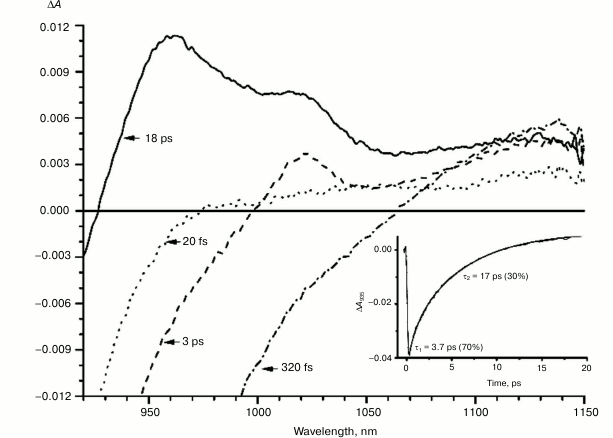
Fig. 3. Absorbance difference spectra of L131LH mutant RCs measured at different delay times. The inset shows photoinduced absorbance changes at 935 nm. The smooth, solid line is the result of fitting the data to a sum of two exponentials. Time constants and amplitudes of exponentials are also shown.
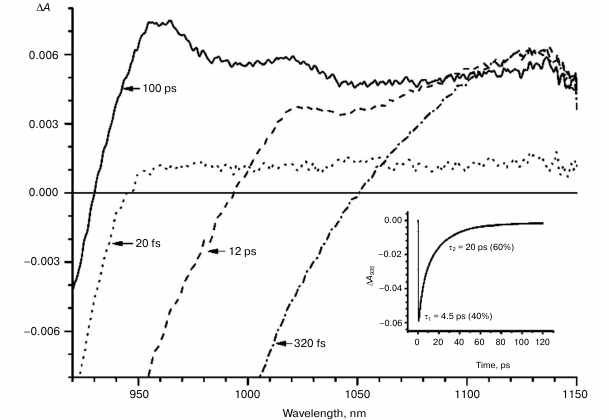
Fig. 4. Absorbance difference spectra of M197FH mutant RCs measured at different delay times. The inset shows photoinduced absorbance changes at 935 nm. The smooth, solid line is the result of fitting the data to a sum of two exponentials. Time constants and amplitudes of exponentials are also shown.
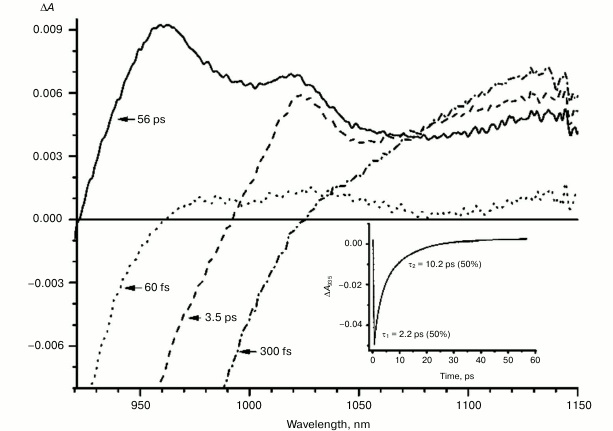
Fig. 5. Absorbance difference spectra of M160LH + L131LH + M197FH mutant RCs measured at different delay times. The inset shows photoinduced absorbance changes at 935 nm. The smooth, solid line is the result of fitting the data to a sum of two exponentials. Time constants and amplitudes of exponentials are also shown.
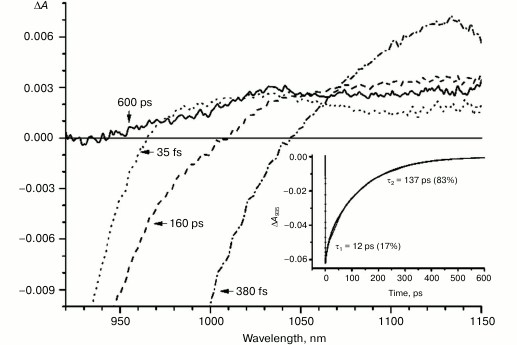
In the triple mutant the formation of the P*(PAδ+PBδ–) state can be monitored by the appearance of an absorption band at >1100 nm. However, the absorption band at 1020 nm is absent at time delays from a few to hundreds of picoseconds, when stimulated emission from P* is already not present (Fig. 5, solid line), and the absorption spectrum at 920-1150 nm does not correspond to the absorption spectrum of the radical cation P+. This suggests that in this mutant the electron does not transfer from P* to the BA molecule or the HA molecule, and the damping of stimulated emission is due to the transition of P* to the ground state. The absence of the absorption of the dimer P+ allows us to track the kinetics of formation and decay of the radical cation band PAδ+ at 1100 nm in the triple mutant (Fig. 6). Absorption in this region, however, does not reach zero even at 600 ps delay, and its origin is unclear.
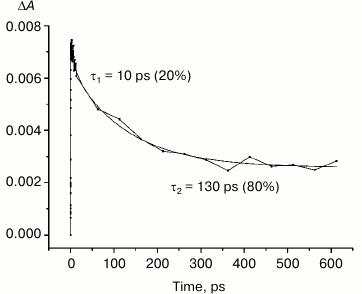
Fig. 6. Photoinduced absorbance changes at 1130 nm measured after excitation of M160LH + L131LH + M197FH mutant RCs at 870 nm with 40-fs at room temperature. The smooth, solid line is the result of fitting the data to a sum of two exponentials. Time constants and amplitudes of exponentials are also shown.
DISCUSSION
Studies and modeling of the electronic structure of the dimer P show that the energy of higher orbitals of monomers PA and PB are different because of the different protein environment. Thus, it is estimated that the energy of higher orbitals of the molecule PB is less than that of PA, which favors the separation of charges in the dimer after the light excitation with the formation of the state P*(PAδ+PBδ–). This difference in the energy of the monomers can be altered by adding hydrogen bonds to one of the molecules. Introduction of a hydrogen bond between the side chain of an amino acid residue (e.g. histidine) and the PB molecule should lead to a reduction in its energy, thus increasing the energy difference between PB and PA, which in turn should increase the likelihood of the charge separation in P*. Hydrogen bonding with the molecule PA should produce the opposite effect, reducing the energetic asymmetry between PA and PB, and thus reducing the probability of charge separation between them. In either case, introduction of hydrogen bonds lowers the energy of the highest molecular orbital of P, increasing redox potential of the primary electron donor. Our results show that the band at 1100 nm is formed in all the investigated reaction centers at approximately the same time with similar amplitude, despite the significant differences in the redox potential of the primary donor P among the mutant RCs. Similarity of the kinetics of the decay of the stimulated emission at 935 nm in the triple mutant (Fig. 5, inset) and decay of absorption in the region of 1100 nm in the same mutant (Fig. 6) indicates that in both cases we observe the same process – the decay of the mixed state P*(PAδ+PBδ–), which is the product of the quantum mechanical and thermodynamic interactions of states, as a result of the return of P* to the ground state, as well as the charge recombination in the PAδ+PBδ– state.
Heterogeneity (the presence of two exponential functions in the approximation) in the kinetics of P* stimulated emission were explained by static heterogeneity of the reaction centers, parallel electron transfer from P* to HA by superexchange, and the reverse transfer reaction P* P+BA– [28-31]. However, recent studies showed little or no dependence of the electron transfer rate on the exciting light wavelength, thus questioning the hypothesis of static heterogeneity [32]. In turn, direct estimate of the electron transfer rate from P* to HA by the superexchange mechanism gives the value of ~1/300 ps, which is much less than the observed rate of transfer of ~1/3 ps [33], allowing us to neglect the transfer by that route. On the other hand, our data (Figs. 1-4, inset) show that heterogeneity of P* stimulated emission kinetics in RCs with single mutations increases together with the increase in the midpoint potential of P. This fact can be interpreted in terms of the reverse transfer model P* P+BA–, i.e. increase in kinetic heterogeneity may originate both from the increase in the reverse electron transfer rate and from the decrease in the direct electron transfer rate due to higher energy level of the P+BA– state relative to the P* state in the mutant RCs. As follows from the figures, the electron transfer in the RCs slows as the redox potential of the primary electron donor P increases, which is reflected in the increase in amplitude of the slow kinetic component.
The mutant RC M197FH is the only one that falls out of this pattern with its potential of P increased by 125 mV, and the rate of the electron transfer to BA being similar to that of the wild type (Figs. 1 and 4, insets). One possible explanation of this phenomenon is the assumption that the replacement of a leucine by histidine at position M197 results not only in increase in the P midpoint potential, but also in increase in the midpoint potential of the radical anion of the monomer bacteriochlorophyll BA. Another possible explanation is reduction of the reorganization energy of the electron transfer reaction or increase in the electronic coupling as a result of the mutation. A similar effect has been described for the mutant RC M197FR with replacement of phenylalanine M197 by arginine. As a result, the midpoint potential of P increased by 80 mV while the rate of electron transfer did not change [34]. The X-ray analysis of the mutant RCs showed that the formation of the hydrogen bond between acetyl carbonyl group of PB and the arginine residue leads to rotation of the group that takes it out of the PB plane, which in turn results in shortening of the distance between the PB and BA molecules. This should lead to an increase in the electronic coupling between PB and BA and, as a result, to acceleration of the electron transfer reaction. The X-ray diffraction analysis also showed that the side chain of the arginine residue in the RC mutant M197FR forms a cavity between P and BA that contains two water molecules donating three hydrogen bonds to the P and BA macrocycles. Such hydrogen bond connection between the electron donor and electron acceptor may also facilitate the tunneling of electrons between the P and BA molecules [34-36]. It is possible that similar effects may be observed in the M197FH mutant RCs.
It is known that the P+BA– energy level lies below the P* energy level by 50-70 mV in native Rba. sphaeroides RCs [14, 37, 38]. Since the redox potential of P in the mutant RCs L131LH is increased by 80 mV compared to native RCs, the energy level of the P+BA– state is expected to be higher than that of the P* state, which should lead to a significant slowing of the electron transfer to the BA molecule. In experiments at low temperature (90 K), the BA– band at 1020 nm in the mutant L131LH RC was not observed [27]. According to our data, the BA– band is clearly observed at room temperature, although its amplitude is lowered. This indicates that the energy gap (ΔG) between the P+BA– and P* states in the L131LH mutant is apparently not much higher than the energy of kT at room temperature (~25 mV), and hence in the wild-type RCs the value of ΔG can be estimated as 60-70 mV.
Figures 1-4, solid line, show that the residual BA– band (10-15% of the maximum amplitude) is present in the difference absorption spectra of the single mutants and the wild-type RCs when the electron transfer to the pheophytin molecule is completed. This can be explained by the presence of thermodynamic equilibrium between the P+BA– and P+HA– states at room temperature. This observation is consistent with the previously proposed model [29, 30], which describes that the energy gap between P* and P+HA– states is small (~350 cm–1) on the time scale of hundreds of picoseconds, and only after hundreds of nanoseconds, as a result of conformational changes of the protein environment accompanied by subsequent relaxation of the radical state, the value of ΔG drops to ~1600 cm–1, which ensures the irreversibility of the reaction.
Thus, in our work experimental evidence of direct participation of the BA molecule in the primary electron transfer in a number of mutant RCs with increased potential of the primary electron donor P was provided. Also data was obtained favoring the electron transfer model in which the energy gap (ΔG) between states P*, P+BA–HA, and P+BAHA– is a value of the same order as the energy of kT at room temperature at time delays of 1-300 ps [31, 39].
This work was supported by a grant from the President of the Russian Federation (NSh-307.2012.4), the grant program of the Presidium of the Russian Academy of Sciences of the IBC, and the Russian Foundation for Basic Research grant (12-04-00332).
REFERENCES
1.Wohri, A. B., Wahlgren, W. Y., Malmerberg, E.,
Johansson, L. C., Neutze, R., and Katona, G. (2009)
Biochemistry, 48, 9831-9838.
2.Stowell, M. H. B., McPhillips, T. M., Rees, D. C.,
Soltis, S. M., Abresch, E., and Feher, G. (1997) Science,
276, 812-816.
3.Khatypov, R. A., Khmelnitskiy, A. Yu., Khristin, A.
M., and Shuvalov, V. A. (2010) Doklady Biochem. Biophys.,
430, 24-28.
4.Khatypov, R. A., Khmelnitskiy, A. Yu., Khristin, A.
M., Fufina, T. Yu., Vasilieva, L. G., and Shuvalov, V. A. (2012)
Biochim. Biophys. Acta, 1817, 1392-1398.
5.Shuvalov, V. A. (1990) Primary Conversion of
Light Energy in Photosynthesis [in Russian], Nauka, Moscow.
6.Kirmaier, C., and Holten, D. (1993) in The
Photosynthetic Reaction Center (Deisenhofer, J., and Norris, J.,
eds.) Academic Press, San Diego, pp. 49-70.
7.Woodbury, N. W., and Allen, J. P. (1995) in
Anoxygenic Photosynthetic Bacteria (Blankenship, R. E., Madigan,
M. T., and Bauer, C. E., eds.) Kluwer Academic Publishers, Dordrecht,
pp. 527-557.
8.Shuvalov, V. A. (2000) Conversion of Light
Energy in Primary Act of Charge Separation in Reaction Centers of
Photosynthesis [in Russian], Nauka, Moscow.
9.Shuvalov, V. A., and Yakovlev, A. G. (2003) FEBS
Lett., 540, 26-34.
10.Deisenhofer, J., Epp, O., Miki, K., Huber, R.,
and Michel, H. (1984) J. Mol. Biol., 180, 385-398.
11.Ermler, U., Fritzsch, G., Buchanan, S. K., and
Michel, H. (1994) Structure, 2, 925-936.
12.Hu, Y., and Mukamel, S. (1990) in Perspectives
in Photosynthesis (Jortner, J., and Pullman, B., eds.) Kluwer
Academic Publishers, Amsterdam, pp. 171-184.
13.Holzapfel, W., Finkele, U., Kaiser, W.,
Oesterhelt, D., Scheer, H., Stilz, H. U., and Zinth, W. (1989) Chem.
Phys. Lett., 160, 1-7.
14.Arlt, T., Schmidt, S., Kaiser, W., Lauterwasser,
C., Meyer, M., Scheer, H., and Zinth, W. (1993) Proc. Natl. Acad.
Sci. USA, 90, 11757-11761.
15.Kennis, J. T., Shkuropatov, A. Y., van Stokkum,
I. H. M., Gast, P., Hoff, A. J., Shuvalov, V. A., and Aartsma, T. J.
(1997) Biochemistry, 36, 16231-16238.
16.Yakovlev, A. G., Shkuropatov, A. Y., and
Shuvalov, V. A. (2000) FEBS Lett., 466, 209-212.
17.Moser, C. C., Keske, J. M., Warncke, K., Farid,
R. S., and Dutton, P. L. (1992) Nature, 355, 796-802.
18.Williams, J. C., Alden, R. G., Murchison, H. A.,
Peloquin, J. M., Woodbury, N. W., and Allen, J. P. (1992)
Biochemistry, 31, 11029-11037.
19.Spiedel, D., Jones, M. R., and Robert, B. (2002)
FEBS Lett., 527, 171-175.
20.Allen, J. P., and Williams, J. C. (1995) J.
Bioenerg. Biomembr., 27, 275-283.
21.Jones, M. R., Visschers, R. W., van Grondelle,
R., and Hunter, C. N. (1992) Biochemistry, 31,
4458-4465.
22.Vasil’eva, L. G., Bolgarina, T. I.,
Khatypov, R. A., Shkuropatov, A. Ya., Miyake, J., and Shuvalov, V. A.
(2001) Doklady Biochem. Biophys., 376, 46-49.
23.Khatypov, R. A., Vasilieva, L. G., Fufina, T. Y.,
Bolgarina, T. I., and Shuvalov, V. A. (2005) Biochemistry
(Moscow), 70, 1256-1261.
24.Cohen-Bazire, G., Sistrom, W. R., and Stanier, R.
Y. (1957) J. Cell. Comp. Physiol., 49, 25-68.
25.Shuvalov, V. A., Shkuropatov, A. Ya., Kulakova,
S. M., Ismailov, M. A., and Shkuropatova, V. A. (1986) Biochim.
Biophys. Acta, 849, 337-346.
26.Woodbury, N. W., Becker, M., Middendorf, D., and
Parson, W. W. (1985) Biochemistry, 24, 7516-7521.
27.Fajer, J., Brune, D. C., Davis, M. S., Forman,
A., and Spaulding, L. D. (1975) Proc. Natl. Acad. Sci. USA,
72, 4956-4960.
28.Du, M., Rosenthal, S. J., Xie, X., DiMagno, T.
J., Schmidt, M., Hanson, D. K., Schiffer, M., Norris, J. R., and
Fleming, G. R. (1992) Proc. Natl. Acad. Sci. USA, 89,
8517-8521.
29.Hamm, P., Gray, K. A., Oesterhelt, D., Feick, R.,
Scheer, H., and Zinth, W. (1993) Biochim. Biophys. Acta,
1142, 99-105.
30.Jia, Y., DiMagno, T. J., Chan, C.-K., Wang, Z.,
Du, M., Hanson, D. K., Schiffer, M., Norris, J. R., Fleming, G. R., and
Popov, M. S. (1993) J. Phys. Chem., 97, 13180-13191.
31.Holzwarth, A. R., and Muller, M. G. (1996)
Biochemistry, 35, 11820-11831.
32.Wang, H., Lin, S., and Woodbury, N. W. (2008)
J. Phys. Chem. B, 112, 14296-14301.
33.Yakovlev, A. G., Vasilieva, L. G., Shkuropatov,
A. Y., and Shuvalov, V. A. (2011) Biochemistry (Moscow),
76, 1107-1119.
34.Ridge, J. P., Fyfe, P. K., McAuley, K. E., van
Brederode, M. E., Robert, B., van Grondelle, R., Isaacs, N. W.,
Cogdell, R. J., and Jones, M. R. (2000) Biochem. J., 351,
567-578.
35.De Rege, P., Williams, S., and Therien, M. (1995)
Science, 269, 1409-1413.
36.Yakovlev, A. G., Shkuropatov, A. Y., and
Shuvalov, V. A. (2002) Biochemistry, 41, 14019-14027.
37.Bixon, M., Jortner, J., and Michel-Beyerle, M. E.
(1995) Chem. Phys., 197, 389-404.
38.Shuvalov, V. A., and Yakovlev, A. G. (1998)
Membr. Cell Biol., 15, 563-569.
39.Van Stokkum, I. H. M., Beekman, L. M. P., Jones,
M. R., van Brederode, M. E., and van Grondelle, R. (1997)
Biochemistry, 36, 11360-11368.