Perspectives of Mitochondrial Medicine
D. B. Zorov1,2*, N. K. Isaev1,2, E. Y. Plotnikov1,2, D. N. Silachev1,2, L. D. Zorova2,3, I. B. Pevzner2,4, M. A. Morosanova2,4, S. S. Jankauskas2,4, S. D. Zorov1,2,4, and V. A. Babenko5
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-0338; E-mail: zorov@genebee.msu.su2Institute of Mitoengineering, Lomonosov Moscow State University, 119991 Moscow, Russia
3International Laser Center, Lomonosov Moscow State University, 119991 Moscow, Russia
4Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia
5Pirogov Russian National Research Medical University, Ministry of Health and Social Development of Russian Federation, ul. Ostrovityanova 1, 117997 Moscow, Russia
* To whom correspondence should be addressed.
Received May 13, 2013
Mitochondrial medicine was established more than 50 years ago after discovery of the very first pathology caused by impaired mitochondria. Since then, more than 100 mitochondrial pathologies have been discovered. However, the number may be significantly higher if we interpret the term “mitochondrial medicine” more widely and include in these pathologies not only those determined by the genetic apparatus of the nucleus and mitochondria, but also acquired mitochondrial defects of non-genetic nature. Now the main problems of mitochondriology arise from methodology, this being due to studies of mitochondrial activities under different models and conditions that are far from the functioning of mitochondria in a cell, organ, or organism. Controversial behavior of mitochondria (“friends and foes”) to some extent might be explained by their bacterial origin with possible preservation of “egoistic” features peculiar to bacteria. Apparently, for normal mitochondrial functioning it is essential to maintain homeostasis of a number of mitochondrial elements such as mitochondrial DNA structure, membrane potential, and the system of mitochondrial quality control. Abrogation of these elements can cause a number of pathologies that have become subjects of mitochondrial medicine. Some approaches to therapy of mitochondrial pathologies are discussed.
KEY WORDS: mitochondria, mitochondrial diseases, mitochondrial DNA, membrane potential, mitochondrial quality control, mitochondria-targeted antioxidants, bacteria, phenoptosisDOI: 10.1134/S0006297913090034
Abbreviations: mtDNA, mitochondrial DNA; nDNA, nuclear DNA; RIRR, ROS-induced ROS release; ROS, reactive oxygen species.
Mitochondrial medicine has emerged as a branch of medical genetics and
is been quickly progressing along with the progress in matching
genetically determined mitochondrial damage and various pathologies. To
date, knowledge of the average mitochondrial proteome includes 1500
proteins [1], most of which are encoded in the
nucleus, and only a small fraction (13 polypeptides in mammalian
mitochondria) is encoded in mitochondria [2]. About
2/3 of these 1500 proteins carry catalytic functions [3], while others carry either structural or yet
unknown functions. In addition, in the case of various oxidative
stress-mediated impacts, a number of proteins are translocated from the
cytosol to mitochondria [4-14], thereby increasing the size of the mitochondrial
proteome. This set of proteins together with unique lipids and
mitochondrial DNA determine not only the functioning of this organelle,
but also the fate of the cell, organ, and apparently the entire
organism [15]. Until recently, constant
reinforcement of the connection between modifications of the nuclear
and mitochondrial genetic apparatus and progress of pathological
processes has given no significant practical results in terms of
therapy, except for trivial symptomatic pharmacological and other
approaches aimed at alleviation of fatal consequences of genetic
modifications, but not removal of their causes [16-19]. Furthermore, not all the
clinical trials have confirmed the efficiency of such approaches [20]. However, the mere recognition of the need to
consider the concerted functioning of the nuclear and mitochondrial
genomes to explain normal or pathological orientation of biological
processes in mitochondria is a great breakthrough.
MITOCHONDRIOLOGY PROBLEMS CAUSED BY THE DEVELOPMENT OF
METHODOLOGY
Certain excessive modern fascination with genomics, when small changes in the level of gene transcription (in either direction) in a number of pathologies [21-23] become the basis for judgment about changes in metabolism proceeding with the participation of proteins encoded by these genes, does not seem justified. First of all, it results from a purely biochemical understanding of any catalytic process carried out by enzymes whose total activity hardly depends on the transcriptome level, being mainly determined by translation rate and possible posttranslational modification of enzymes. The number of enzymes and their catalytic activity (it is these characteristics that determine the norm or pathology) depend on many factors, including low and high molecular weight regulators of enzymatic activity, and it is these factors that are more important in discussing the nature of pathologies than the effects of the transcription level of different genes. Certain imbalance towards the role of transcription in the development of pathologies (including the aging process [24, 25]) is perhaps historically justified until the time when enzymatic processes are studied in situ, and then also in vivo. The current situation, when this mission still cannot be carried out, does not justify such an imbalance towards the methods available today, these producing a simplified molecular-biological picture. It rather calls for speeding the development of a new methodology of fine registration of biochemical processes while maintaining all the regulatory elements intact in living cells, tissues, and organisms.
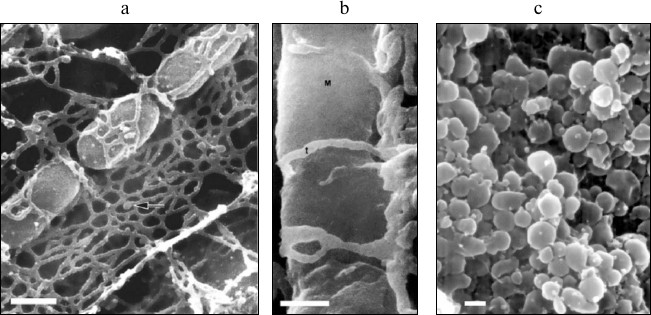
Modern biology has undoubtedly made a great breakthrough in the transition from a “complex” biological system to a “simpler” one (organism–organ–cell–organelle–molecule). The object of study has naturally lost its dependence on the structural–functional organization of the organism in the course of this transition. Let us present just one example of the complex structural organization of mitochondria in cells – photographs obtained by scanning electron microscopy (Fig. 1) [26-28]. It is clear from these photographs that structural interaction between mitochondrial and reticular cellular structures is so pronounced that a methodological shift from an isolated cell to an isolated mitochondrion inevitably leads to a loss of the vital qualities of mitochondria characteristic of living cells. These structural interactions of two intracellular compartments [29] gave rise to a theory of their functional interaction [30-32], which is also lost due to methodological separation of these structures.
Fig. 1. Scanning electron microscopy of mitochondria in the tissue (cardiomyocytes of a dog; arrow indicates the elements of sarcoplasmic reticulum [26] (a); lateral vastus; t, transverse tubules; M, mitochondrion [27] (b)) and of isolated mitochondria of monkey heart (c) [28]. The scale is 0.1 µm. Reprinted with the permission of the authors.
It should be noted that not only a cell–organelle transition (as shown for the mitochondria–reticulum interaction) is essential for the reflection of the real functioning of these two systems, but also the transition from an isolated organ, or, at least, from a multicomponent mixture of organ cells to individual cells of the same type, turns out to be partially defective. The latter can be first of all explained by the fact that, for example, a pure neuronal culture (and that is the goal of neurocytologists – not to allow a substantial “contamination” of a neuronal cell culture with the cells of non-neuronal nature, such as glia) does not exist in an organism. Brain neurons are known to maintain their activity largely due to interactions with non-neuronal cells (astrocytes, microglia, and oligodendrocytes in the central nervous system; Schwann cells in the peripheral nervous system) [33]. Earlier, astrocytes were thought to fulfill a rather passive role, while now it has become obvious that they directly participate in synaptic transmission [34, 35], control of cerebral blood flow [36], development [37], formation of neural networks [38], etc. These facts are enough to suggest the thoroughly purified from glia culture of neurons does not adequately reflect (in a functional sense) the processes that neurobiologists seek to understand.
This logic is fully applicable to other organs formed of cells different in structure and function (kidney, heart, etc.). One needs to understand that organs are formed due to not only peaceful, but also mutually beneficial coexistence of different cell types, and individual functioning of each cell type can vary greatly from their operation in a community. The latter determines a clear separation of functions for each fraction and causes specialization leading to spatial and functional integration through different signals.
This leads us to the understanding of the need to develop approaches that would allow studying cells in organs, instead of limiting work to research on pure cell cultures.
If we continue the same line of reasoning along the ascending complexity of the organization of biological systems, we will see that there are many data on inter-organ signaling, when functioning of a particular organ is impossible without signals coming from other organs. Twenty years ago the term “remote preconditioning” has been established, a phenomenon leading to the reduction of damage induced in a primary organ (heart or brain) due to preconditioning of a secondary organ (kidney or muscle) [39-42].
Great effort is now needed to summarize the data obtained at molecular biological level so as to fulfill the pressing demand of making the reverse transition of studies of biological objects from a simpler to more complex organization (ideally, molecule–organelle–cell–organ–organism). First of all, these are the requirements of modern biochemistry and physiology, which study pathological processes so as to develop methods of pharmacological treatment intended to normalize the activity of an organism suffering from such pathologies. This logic is applicable to any pathology including those caused by improper functioning of mitochondria.
ORIGINS OF MITOCHONDRIAL MEDICINE
Historically, the pathology that was later named after one of the researchers is considered to be the first declared mitochondrial disease. It was discovered in clinical practice more than half a century ago. This disease of a 30-year-old woman was characterized by extremely severe hypermetabolism not related to hyperthyroidism [43]. The discovery of this disease by endocrinologist Rolf Luft coincided with the beginning of the flourishing of world bioenergetics, and Luft cooperated with one of the founding fathers of this science, Lars Ernster [44]. In the Ernster laboratory, this patient’s problem was found to be caused by her skeletal muscle mitochondria, which lost control over respiratory oxidative phosphorylation, resulting in high energy loss; the biological efficiency of energetics (in terms of ATP synthesis level) was shown to be extremely low.
Thus, it was over 50 years ago that a new type of medicine started – mitochondrial medicine – based on pathologies caused by improper functioning of mitochondria. There is no accurate assessment of the human population with mitochondrial diseases in the world, but, for example, the assessment of this parameter in the north-east of England showed that there is one mitochondrial patient with pronounced clinical manifestations per 10,000 people and one patient with probable development of mitochondrial disease per 6000 people [45]. The most pessimistic estimate of pathogenic mutations in mtDNA is one case per 200 people [46, 47]. Description of all discovered pathogenic mtDNA mutations can be found in the database MitoMap (http://www.mitomap.org).
MITOCHONDRIA – FRIENDS OR ENEMIES?
The question of mitochondria being friends or enemies seems to be absurd, as even school textbooks teach mitochondria to be endosymbionts. However, more and more information on extremely “egoistic” behavior of mitochondria, largely subjugating cell metabolism to their own needs, is being accumulated. We will discuss some details of this possible “egoism” when discussing mitochondrial homeostasis; as a result, it will be more difficult to speak about the obviousness of endosymbiotic coexistence.
In the case of ancient primitive eukaryotes, mitochondria were pronounced intracellular parasites of bacterial origin that moved part of their genome to the nucleus in the course of evolution [48]. As a result, mitochondria became totally dependent on the functioning of the nucleus; however, they preserved some genetic control points in their own, albeit reduced genome (mtDNA) discovered in 1963 [49]. Pathogenic deletions and point mutations of mtDNA, the basis of mitochondrial diseases, were discovered 25 years later [50, 51]. Among them we can list such pathologies as Leigh syndrome or Leigh disease (subacute necrotizing encephalomyopathy), syndrome of mitochondrial encephalopathy with lactic acidosis and insult-like episodes, syndrome of myoclonic epilepsy with ragged-red fibers, etc. The genetic basis of these diseases is quite obvious. We will not consider them in detail as there are many good reviews on this topic (for example, [52, 53]).
Genetic defects causing abnormal functioning of mitochondrial structures are still technically unrepairable. However, it should be understood that knowledge obtained due to mitochondriology can help to diagnose mitochondrial diseases. This was elegantly used at one point by Britton Chance, who showed how knowledge of fundamental bioenergetics can help in the treatment of genetic pathologies. A seventeen-year-old patient restricted to a wheelchair because of muscular weakness and lactic acidosis due to a genetic defect of respiratory chain complex III, which caused myoclonic epilepsy with ragged-red fibers, partially recovered after taking a mixture of ascorbate and menadione, shunting the affected area of the respiratory chain [54]. This simple procedure of course did not abolish the genetic defect, but it provided electron transport along the respiratory chain coupled with moderate ATP synthesis, which partially restored muscle activity.
Phenotypic expression of an mtDNA genetic defect depends on the level of heteroplasmy of the affected mtDNA chain with a complementary wild-type chain. The unclear nature of segregation of mtDNA plays a huge role in this process [55]. Each phenotypic expression of mitochondrial pathology has its own characteristic threshold level of heteroplasmy, which, according to various estimates, is about 60% for mDNA deletions and about 90% for other mutations [56]. Straight-line logic requires trying to prevent reaching phenotypic threshold due to maximal possible dilution of the “wrong” mtDNA copy with the wild-type [52, 57, 58].
Instability of mtDNA is one of the main causes of mitochondrial diseases, and it is probably the main target of mitochondrial medicine, which, as we have noted, is still in its infancy. However, Nature herself suggests ways that should be followed to find a strategy for therapy of mitochondrial diseases.
One of the ways used by nature to eliminate mtDNA defects is mtDNA repair, a process whose elements similar to those in nuclear DNA have been described in a number of studies [59, 60].
The second, more radical way to fight the “wrong” mtDNA copies is to destroy these copies together with their own “home”, that is, with the mitochondrion. This process was discovered 30 years ago – the global fragmentation of the mitochondrial population [61] (other terms – fission of filamentous mitochondria [62] and their “thread–grain” transition [63]). This phenomenon is obviously a basic process of mitochondrial evolution, which probably determines the mechanism of mtDNA segregation. Simple logic suggests the unified mitochondrial system (usually represented in cells by a single elongated, often branched mitochondriome [64, 65]) to be rational only in an optimal and healthy environment. In the case of pathologies, first of all those coupled to high level of oxidative stress, one can observe global fragmentation of the mitochondrial reticulum [61, 66, 67], apparently due to the necessity of natural fractioning of mitochondria within one cell separating a fraction to be eliminated. It has to be kept in mind that there are many copies of mtDNA in an ordinary healthy cell, the number of copies varying from ~1000 in regular somatic cells to 100,000 in oocytes [68]. Purely theoretically, only one mtDNA copy can be present in each mitochondrion in the process of forced global mitochondria fragmentation (or may not be present at all), but in practice it is very difficult to make such a calculation. It is obvious that mitochondrial fragments with genetically defective mtDNA (especially those with a single mtDNA copy) will not be able to provide the proper functioning of mitochondria, first of all the work of respiratory proton pumps and ATP-synthase complex. By definition, such mitochondria may be unable to maintain their membrane potential by the proton pumping (if mtDNA defects affect complexes I, II, III, or IV of the respiratory chain) or due to the reversal of the ATP-synthase reaction (if the defect affects complex V). Such “wrong” mitochondria should be eliminated, low mitochondrial membrane potential probably being the main criterion for the selection (the role of mitochondrial membrane potential will be discussed separately). Under laboratory conditions, there are many approaches to isolation of populations of homologous and heterologous cells with different mitochondrial membrane potentials for further identification and characterization of cells [69].
In recent years, the concept of elimination of low-potential mitochondria via so-called autophagy (translated from Greek “self-eating”) has been thoroughly confirmed. This phenomenon is now called mitophagy, a process known for quite a long time and described in old studies on lysosomal degradation of mitochondria [70, 71]. Mitochondria are almost completely digested in lysosomes. Interestingly, lysosome extracts do not hydrolyze one of the most important mitochondrial lipids, cardiolipin, which may be the cause of its immunogenicity in syphilis, the diagnostic basis of the Wassermann–Neisser–Brucke reaction [72].
Detouring from the main topic, we would like to note that anticardiolipin antibodies were the first discovered of nine groups of mitochondrial autoantibodies circulating in blood in different diseases (primary biliary cirrhosis, congestive heart failure, toxic hepatitis, toxic pseudo-lupus, etc. [73, 74]), which also need to be classified as mitochondrial diseases. The reason for mitochondrial antigens (which should obviously be destroyed along with mitochondria) being present in blood remains unclear. The very process of immune response to mitochondrial derivatives via antibody formation is also mysterious as the classical immune system of an organism is considered [75] to be tolerant to the constituent parts of its own mitochondria while responding to similar, related bacterial elements. A completely different situation arises in the activation of innate immunity, which, through a toll-like receptors, probably equally responds to both mitochondrial and related bacterial antigens. One possible explanation is that it is still unclear whether recently discovered DAMPs (damage-associated molecular patterns) [76] are a set of intact mitochondrial components or the damage causing their appearance somewhat modifies these components, resulting in the innate immune system reacting to them. The latter has been, for example, shown for oxidized mtDNA, which is directly involved in activation of inflammasomes [77].
It should be noted that mitochondrial pathophysiology is rightfully to be considered much more broadly than was declared 50 years ago, now covering not only genetically inherited but also acquired mitochondrial defects, which, of course, affect the cellular genetic apparatus. There is an urgent need for a broader interpretation of the term “mitochondrial medicine” due to the fact that earlier this term was largely restricted to mtDNA genetic rearrangements that resulted in pathologies, while now there are many other phenomena that can be implicated in it. These phenomena embrace all the violations of the mitochondrial proteome caused by the combined processing of mitochondrial and nuclear DNA, which leads to the changes in transcription, translation, and posttranslational modification of all the proteins present in mitochondria at any moment of their life cycle. These modifications surely affect all the mitochondrial content: proteins, lipids, nucleic acids, and metabolites.
Concerted action of the nuclear and mitochondrial genomes for the functioning of mitochondria while maintaining the key control elements within the mitochondria requires maximal and precise quality of the control. The mitochondrial life cycle is not too long, probably due to the aggressive chemical environment of mitochondrial content, which leads to constant damages in the structure of mitochondrial components, primarily related to redox transformations. Systems of mtDNA repair obviously fail to ensure the complete elimination of oxidative damage of the mitochondrial genome, which is extraordinarily high and exceeds that of the nuclear genome 10-20-fold [78, 79].
The quality of the mitochondrial population is controlled in two ways. On one hand, it is a constant process of fusion and fission of mitochondria in cells where it is possible, leading to the “mixing” of mitochondria in a cell, possibly accompanied by “mixing” of their genetic content and segregation of mtDNA. On the other hand, as we have already mentioned, organelles that have accumulated “errors” are physically and chemically destroyed.
MULTIFACETED ROLE OF MITOCHONDRIAL MEMBRANE POTENTIAL
Mitochondrial membrane potential (Δψ), which is the major part of the transmembrane proton potential in an animal cell, is the driving force for ATP synthesis in the organelle [80-82]. We mentioned above and previously [83, 84] that membrane potential is the critical factor determining mitochondrial viability, and mitochondria with membrane potential values not optimal for cellular processes are eliminated. In particular, this is needed to avoid even the minimal possibility of uncontrolled triggering of unwanted cell killing by mitochondria [83, 85, 86]. This is why in terms of energy consumption a very “expensive” system of mitochondrial quality control is constantly at work in cells. Poor quality mitochondria and their components are subject to non-lysosomal degradation, which starts with specific labeling by ubiquitination followed by sending labeled mitochondria to the system of proteasomal degradation [87-89].
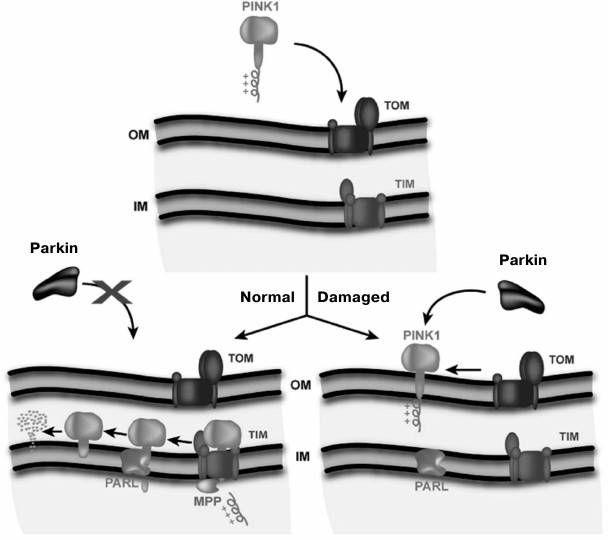
Elimination of mitochondria with low membrane potential in neurons characteristic for Parkinson’s disease follows an elegant scheme proposed by Youle et al. [90] (Fig. 2). According to this scheme, the protein parkin is present in the cytoplasm of the neuron. Parkin, an E3 ubiquitin lyase, uses a kinase PINK1 (which carries a mitochondrial address) also present in the cytosol as its substrate. The full length PINK1 is rapidly transported into the mitochondrial matrix when potential in mitochondria is high. Along the way, MPP peptidase cleaves the signal peptide from PINK1. As a result, there is practically no PINK1 in the cytosol. Once in the matrix, this kinase is immediately degraded by the protease PARL. In case of low mitochondrial membrane potential, PINK1 is only anchored in the outer mitochondrial membrane and is not transported into the matrix; because of this, PINK1 becomes an easy prey for parkin to be ubiquitinated. As a result, the mitochondrion becomes sentenced to proteasomal degradation.
Fig. 2. Model of the regulation of mitochondrial quality by membrane potential (Δψ). TOM, the system of protein transport across the outer mitochondrial membrane (OM); TIM, the system of transport across the inner membrane (IM). Healthy mitochondria are shown on the left side, and damaged mitochondria unable to generate sufficient membrane potential on the right side (Jin et al., 2010 [90]. Scheme originally published in J. Cell Biol., DOI: 10.1083/jcb.201008084).
The process controlling the quality of mitochondria is one of the “hottest” research topics for molecular and cell biologists today. It has become absolutely clear that violation of this process is one of the causes of a number of pathologies and is directly related to mitochondrial medicine. It is also becoming clear that the quality of mitochondria depends on the membrane potential value on the inner mitochondrial membrane, which is monitored by mitochondrial quality control systems similar to the above-described parkin system, and homeostasis of the mitochondrial membrane potential is one of the indispensable foundations of mitochondrial therapy. Maintenance of membrane potential is known to be absolutely crucial for mitochondria because even at a time when mitochondria are unable to generate Δψ by proton pumps (e.g. at anoxia), mitochondria start consuming cellular ATP by reversal of the mitochondrial ATP-synthase to maintain Δψ [91].
The maintenance of membrane potential at the expense of cellular ATP illustrates the mentioned above “egoistic” behavior of mitochondria. It is quite obvious that ATP-based electrical charge on the mitochondrial membrane is a serious difference between mitochondria and their free-living bacterial ancestors, which could not afford such a process as there is no ATP available in their unlimited aqueous environment. Therefore, bacteria do not need such a transport protein as the ATP/ADP translocator, which, by definition, is an ADP supplier, traveling from the cytoplasm into the mitochondrial matrix to the ATP synthase complex. Let us note that nucleotide exchange at the translocator is an electrogenic process, and high membrane potential will slow ATP uptake into the matrix because ATP has a higher negative charge than ADP. Thus, the situation changes in the case of parasitic existence in a closed and very limited space (inside the cell), where ATP level is sufficient to support a number of cellular processes.
A paradoxically high level of intracellular ATP remains a mystery though, as it significantly exceeds the kinetic constants of all the ATPases and leads to full saturation of intracellular enzymes with ATP and complete absence of a control from ATP levels. This can be understood if we assume mitochondria to produce so high ATP primarily for their own needs – to buffer their membrane potential and ensure its homeostasis, which at least temporarily would not depend on the supply of substrates. The fact that the cell can use this ATP for its non-mitochondrial needs is fully compensated by mitochondria utilizing acetyl-CoA, which is used in the tricarboxylic acid cycle or in fatty acids synthesis. Here we can see a very fine line between mitochondrial parasitism (egoism) and symbiosis between mitochondria and other cell compartments. This assumption creates a basis for understanding one of the indispensable foundations of mitochondrial medicine, determining the need for homeostasis of the mitochondrial membrane potential, i.e. keeping it at a certain high enough level, the loss of which causes pathologies.
Let us briefly consider the complicated role of mitochondrial membrane potential in bioenergetics. There is a certain “window” in mitochondrial membrane potential values when thermodynamic considerations allow for the synthesis of a high-energy phosphate bond in the ATP molecule [92]. High levels of membrane potential are quite desirable from the perspective of energy reserves and reliability, but undesirable from the perspective of the possible production of an excessive level of reactive oxygen species (ROS) coupled to these high values [93, 94]. It is clear that the bioenergetic function of mitochondria is realized within this “window”, with a possible compromise between bioenergetic demands and production of signaling and pathogenic molecules. Given the existence of this window, and based on the pathogenicity of ROS produced in the mitochondrial respiratory chain, it was suggested that the efficiency of a bioenergetic machine should be somewhat weakened artificially so as to reduce ROS production by reducing membrane potential to the threshold level when ATP synthesis is still possible. This can be achieved either by not very effective uncouplers of oxidative phosphorylation or by low concentrations of effective uncouplers, in both cases achieving the effect of “mild” uncoupling as one of the possible therapeutic approaches used in mitochondrial medicine [93, 95-97]. It should be noted that “mild” uncoupling will also inevitably lead to a certain decrease in cellular ATP level and a subsequent increase in the level of AMP, which activates AMP-dependent protein kinase (AMPK). Activation of this kinase leads to the launch of the correction mechanisms in a number of pathologies, such as diabetes [98]. Restriction of consumed calories, which is believed to slow the aging process, also leads to activation of AMPK [99] coupled to a sharp reduction in ATP level. We can also conclude that in contrast to mitochondrial Δψ, the change in cellular ATP homeostasis is likely not to trigger pathologies, but rather the opposite.
There is also another perspective on the critical role of ATP in a cell where its level is constantly monitored by different systems [100], but perhaps it is the radical drop in ATP level below certain critical values that is extremely undesirable (pathological). When such a threshold is reached, the cardiomyocyte is known to enter the state of rigor. Hence, similar to Δψ, in this case there is also a “window” of acceptable intracellular ATP concentration, within which a small drop in this level can be advantageous. If we take into account the above-defined requirement of membrane potential homeostasis as one of the prerequisites for preventing mitochondrial pathologies, this requirement should be reconciled with approaches causing “mild” uncoupling. Nevertheless, perhaps membrane potential changes within the above-mentioned acceptable “window” can satisfy this requirement. Whether “mild” uncoupling is a therapeutically justified approach, reducing the risk of pathological processes, remains to be seen, as the debate on this issue continues [101].
In addition to stable and long lasting Δψ changes caused by uncouplers, transient Δψ oscillations can be observed in mitochondria of cardiomyocytes exposed to a local or global oxidative stress. These oscillations often accompanied by a sharp increase in ROS generation caused by prolonged exposure to low doses of ROS (ROS-induced ROS release (RIRR)) [102-104]. Oscillations spread in three-dimensional space along the chain of mitochondria connected by electrical contacts [64, 65], starting from a single mitochondrion as the primary oscillator and finishing with the oscillation of the entire mitochondrial population, which is not always synchronized. Analysis of these oscillations led to the conclusion of their pathological nature and direction. Oscillations in mitochondrial membrane potential were found to be coupled not only to oscillations of mitochondrial redox components (pyridine and flavin nucleotides [105]), but also to fluctuations in ion currents through sarcolemma causing shortening and suppression of action potential on the cardiomyocyte cell membrane, which is directly connected to such pathologies as heart arrhythmias [106]. ROS, as the main pathogenic agents, were declared to be the primary elements triggering oscillations of mitochondrial Δψ [102, 107]. The critical threshold for triggering RIRR, being accompanied by generation of a mitochondrial pore (nonspecific permeability) [102] or an anion channel in the inner membrane [108], also limits mitochondrial resistance to the effects of an introduced oxidant. This resistance threshold in response to external ROS was given the name “mitochondrial criticality”; it means reaching the level when conditions for Δψ oscillations are created. The latter, in turn, creates a temporal and spatial heterogeneity of excitability in the heart [109, 110], causing cardiac arrhythmias, which often end in cardiac arrest and death of the individual. High threshold of RIRR induction is the basis of durability and capacity of the mitochondrial redox buffer, the rate of its use being determined by the level of external oxidant introduced to mitochondria [9]. Ligands of the mitochondrial benzodiazepine channel eliminated both RIRR-induced Δψ oscillations and ventricular tachycardia and fibrillation [111], thereby placing mitochondrial benzodiazepine receptor [61, 112, 113] in a set of important targets for the therapy of certain diseases mediated by mitochondrial dysfunction. Superoxide dismutase mimetics and mitochondria-targeted antioxidant SkQ1 showed similar antiarrhythmic effect, suggesting antioxidant therapy to be a potential solution to some problems of mitochondrial medicine [111, 114].
CONTROVERSIAL ISSUES CONCERNING THE USE OF ANTIOXIDANTS AS
THERAPEUTIC AGENTS
In recent years criticism has arisen of researchers proposing to treat various pathologies, including mitochondrial disorders, with antioxidants. Such criticism of scientists trying to fight adverse effects of oxidative stress is stemmed from the fact that various antioxidants showed the full range of both positive and negative effects in a series of clinical trials [115-119]. It is obviously possible for natural antioxidant vitamins (known for their efficiency) to act differently than their synthetic analogs used in clinic [120, 121], so copying of the natural products does not seem to be very successful. It also means that not all antioxidants have equal therapeutic potency. The latter may reflect the fact that not only the antioxidative part is essential in the antioxidant molecule, but also another one, which carries out a number of non-antioxidative functions [122]. As a result, reasonable point of view begins to prevail that indiscriminate recognition of oxidants as sources of pathogenesis should be abandoned. However, this does not mean that we should abandon the use of antioxidants in the treatment of some diseases.
A popular opinion of stress having a healing power does not seem to fit into rigorous theory and practice of oxidative damage to cellular components, but it can also be explained primarily by the fact that today the concept of oxidative stress is very relative, and there is no quantitative assessment of its level. Furthermore, the heterogeneity of ROS generation levels in different cells (compare, for example, macrophages or alveolar cells and fibroblasts) in the population of the same cells (closer or further away from the blood vessel) or in the population of mitochondria inhabiting the same cell (for example, subsarcolemmal and interfibrillar mitochondria in a muscle cell) make the notion of oxidative stress even more vague. Some averaging of the level of oxidant distribution will not produce impressive results and will not help to come up with objective parameters for quantitative evaluation of the level of oxidative stress. However, it is quite important that we have come to the understanding of the necessity of an optimal (even averaged) oxidant level, which is essential for the fulfillment of the vast range of cellular functions. It is unclear where to draw the line between pathological and non-pathological (if it exists) oxidative stress, but we can assume a controlled, small excess of oxidants above the normal level triggers a series of processes aimed at elimination of this increase and activation of a number of processes useful for the cell, organ, and the entire organism, which will lead to the renewal, increase in resistance to pathological factors, and stability of the biological system following a feedback mechanism. (It is quite possible that this mechanism is implemented in the tissues of the naked mole-rat, which is a surprisingly long-lived animal while having clearly elevated levels of oxidants and oxidative modifications [123].) Following the same mechanism of feedback, antioxidants can cause unwanted sharp increase in oxidant generation in response to its artificial suppression, which will be seen as a prooxidant effect executed by an antioxidant.
The situation looks very different when generation of oxidants fails to be strictly controlled [102]. It results in pathogenic uncontrolled oxidation of vital cellular components, which triggers pathological changes in an organ and entire organism, which often proves to be fatal for the latter. In this case, the use of antioxidants is theoretically and practically justified, and there are many such examples.
An anti-phenoptotic effect of mitochondria-targeted antioxidants demonstrated in our group [15] is a unique example that confirms the validity of the arguments presented above. We managed to distinguish between nephroprotective and anti-phenoptotic effects using two mitochondria-targeted antioxidants (SkQ1 and SkQR1) [124, 125]. While only one antioxidant (SkQR1) showed nephroprotection from the harmful effect coupled to oxidative stress, both of them equally protected animals from death caused by renal dysfunction [114, 126]. The impression was that, first, the targets ultimately providing the protection of the organ and the organism are different, and, second, the antioxidative effect of the molecule of the mitochondria-targeted antioxidant could be sufficient for the protection of the organism, while another effect of an unidentified mechanism, inherent only to SkQR1, was needed for the organ protection (in addition to antioxidative effect). We confirmed the selectivity of antioxidant effects and the necessity to select therapeutic efficiency of all the antioxidants, including mitochondria-targeted ones. Later we showed that administration of SkQR1 mobilizes the defense mechanisms of an animal, in particular, by inducing the synthesis of its universal “defender”, erythropoietin [127], which triggers a cascade of anti-ischemic protection. This confirms the possibility of positive non-antioxidant effect of a number of antioxidants that might be used successfully. We conclude that antioxidants of both narrow and wide action can and should be studied so as to understand their ability to prevent phenoptosis of various natures, by affecting mitochondrial functioning.
NEED TO UNDERSTAND THE BACTERIAL NATURE OF MITOCHONDRIA FOR
DEVELOPMENT OF THE STRATEGY OF MITOCHONDRIAL MEDICINE
Previously, we examined in detail the problem of general similarity between mitochondria and bacteria, which clearly points to a common ancestor [15]. As part of the description of the possible mechanisms of phenoptosis or multiple organ failure, we have already considered the question of the pathogenic role of mitochondrial fragments (DAMPs) [76], which can be released into the blood stream and cause immune response similar to that caused by entire bacteria and their components. This immune response (septic in case of bacteria and sepsis-like in the case of mitochondria [128, 129]) can often be the cause of the death of an organism [130]. As a result, we assume mitochondrion to be that generator that triggers programmed death in many cases [15].
This understanding, first of all, puts immunology, as a part of mitochondrial medicine, among the most important disciplines, the results of which will determine the strategy for mortality prevention. Second, it requires constant consideration of the bacterial origin of mitochondria and their present similarity to bacterial “relatives”. This is needed so as to compare the strategies of antibacterial and anti-phenoptotic protection for the development of a universal strategy to fight death possibly caused by mitochondrial dysfunctions. The key points of such a strategy may include the maintenance of mtDNA homeostasis, regulation of mitochondrial membrane potential, and maintenance of mitochondrial quality control systems with all their elements including also the destruction of damaged mitochondrial structures. A broad range of activities is needed to implement such a strategy, in particular, pharmacological correction of oxidative stress within reasonable limits in the presence of oxidative and other modifications of mitochondrial components responsible for the maintenance of the mentioned types of homeostasis.
This work was supported by Russian Foundation for Basic Research (grants 1-04-01307 (DBZ), 12-04-00025 (NKI), 11-04-00771 (EYP), 13-04-00484 (DNS)) and a grant of the President of the Russian Federation (MK-729.2012.4 (DNS)).
REFERENCES
1.Lopez, M. F., Kristal, B. S., Chernokalskaya, E.,
Lazarev, A., Shestopalov, A. I., Bogdanova, A., and Robinson, M. (2000)
Electrophoresis, 21, 3427-3440.
2.Anderson, S., Bankier, A. T., Barrell, B. G., de
Bruijn, M. H., Coulson, A. R., Drouin, J., Eperon, I. C., Nierlich, D.
P., Roe, B. A., Sanger, F., Schreier, P. H., Smith, A. J., Staden, R.,
and Young, I. G. (1981) Nature, 290, 457-465.
3.Naviaux, R. K. (2012) J. Pharmacol. Exp.
Ther., 342, 608-618.
4.Ahmed, S., Passos, J. F., Birket, M. J., Beckmann,
T., Brings, S., Peters, H., Birch-Machin, M. A., von Zglinicki, T., and
Saretzki, G. (2008) J. Cell Sci., 121, 1046-1053.
5.Chen, J., and Siddiqui, A. (2007) J. Virol.,
81, 6757-6760.
6.Cuttle, L., Zhang, X. J., Endre, Z. H., Winterford,
C., and Gobe, G. C. (2001) Kidney Int., 59,
1779-1788.
7.Gross, A., Yin, X. M., Wang, K., Wei, M. C.,
Jockel, J., Milliman, C., Erdjument-Bromage, H., Tempst, P., and
Korsmeyer, S. J. (1999) J. Biol. Chem., 274,
1156-1163.
8.Heo, J. M., Livnat-Levanon, N., Taylor, E. B.,
Jones, K. T., Dephoure, N., Ring, J., Xie, J., Brodsky, J. L., Madeo,
F., Gygi, S. P., Ashrafi, K., Glickman, M. H., and Rutter, J. (2010)
Mol. Cell, 40, 465-480.
9.Juhaszova, M., Zorov, D. B., Kim, S. H., Pepe, S.,
Fu, Q., Fishbein, K. W., Ziman, B. D., Wang, S., Ytrehus, K., Antos, C.
L., Olson, E. N., and Sollott, S. J. (2004) J. Clin. Invest.,
113, 1535-1549.
10.Majumder, P. K., Mishra, N. C., Sun, X., Bharti,
A., Kharbanda, S., Saxena, S., and Kufe, D. (2001) Cell Growth
Differ., 12, 465-470.
11.Nomura, M., Shimizu, S., Ito, T., Narita, M.,
Matsuda, H., and Tsujimoto, Y. (1999) Cancer Res., 59,
5542-5548.
12.Qi, X., Disatnik, M. H., Shen, N., Sobel, R. A.,
and Mochly-Rosen, D. (2010) Mol. Biol. Cell, 22,
256-265.
13.Tombal, B., Weeraratna, A. T., Denmeade, S. R.,
and Isaacs, J. T. (2000) Prostate, 43, 303-317.
14.Zhao, Y., Chaiswing, L., Velez, J. M.,
Batinic-Haberle, I., Colburn, N. H., Oberley, T. D., and St. Clair, D.
K. (2005) Cancer Res., 65, 3745-3750.
15.Zorov, D. B., Plotnikov, E. Y., Jankauskas, S.
S., Isaev, N. K., Silachev, D. N., Zorova, L. D., Pevzner, I. B.,
Pulkova, N. V., Zorov, S. D., and Morosanova, M. A. (2012)
Biochemistry (Moscow), 77, 742-753.
16.Schmelzer, C., and Doring, F. (2012) Mutat.
Res., 733, 61-68.
17.Klopstock, T., Yu-Wai-Man, P., Dimitriadis, K.,
Rouleau, J., Heck, S., Bailie, M., Atawan, A., Chattopadhyay, S.,
Schubert, M., Garip, A., Kernt, M., Petraki, D., Rummey, C., Leinonen,
M., Metz, G., Griffiths, P. G., Meier, T., and Chinnery, P. F. (2011)
Brain, 134, 2677-2686.
18.Jeppesen, T. D., Schwartz, M., Olsen, D. B.,
Wibrand, F., Krag, T., Duno, M., Hauerslev, S., and Vissing, J. (2006)
Brain, 129, 3402-3412.
19.Taivassalo, T., Gardner, J. L., Taylor, R. W.,
Schaefer, A. M., Newman, J., Barron, M. J., Haller, R. G., and
Turnbull, D. M. (2006) Brain, 129, 3391-3401.
20.Pfeffer, G., Majamaa, K., Turnbull, D. M.,
Thorburn, D., and Chinnery, P. F. (2012) Cochrane Database Syst.
Rev., 4, CD004426.
21.Schena, M., Shalon, D., Davis, R. W., and Brown,
P. O. (1995) Science, 270, 467-470.
22.Kittleson, M. M., Minhas, K. M., Irizarry, R. A.,
Ye, S. Q., Edness, G., Breton, E., Conte, J. V., Tomaselli, G., Garcia,
J. G., and Hare, J. M. (2005) Physiol. Genom., 21,
299-307.
23.Cooper-Knock, J., Kirby, J., Ferraiuolo, L.,
Heath, P. R., Rattray, M., and Shaw, P. J. (2012) Nat. Rev.
Neurol., 8, 518-530.
24.Sheydina, A., Volkova, M., Jiang, L., Juhasz, O.,
Zhang, J., Tae, H. J., Perino, M. G., Wang, M., Zhu, Y., Lakatta, E.
G., and Boheler, K. R. (2012) Aging. Cell, 11,
350-359.
25.Zahn, J. M., Poosala, S., Owen, A. B., Ingram, D.
K., Lustig, A., Carter, A., Weeraratna, A. T., Taub, D. D., Gorospe,
M., Mazan-Mamczarz, K., Lakatta, E. G., Boheler, K. R., Xu, X.,
Mattson, M. P., Falco, G., Ko, M. S., Schlessinger, D., Firman, J.,
Kummerfeld, S. K., Wood, W. H., 3rd, Zonderman, A. B., Kim, S. K., and
Becker, K. G. (2007) PLoS Genet., 3, e201.
26.Yoshikane, H., Nihei, T., and Moriyama, K. (1986)
J. Submicrosc. Cytol., 18, 629-636.
27.Ogata, T., and Yamasaki, Y. (1987) Cell Tissue
Res., 250, 489-497.
28.Shimada, T., Morizono, T., Yoshimura, T.,
Murakami, M., and Ogura, R. (1978) J. Electron. Microsc.
(Tokyo), 27, 207-213.
29.Helle, S. C., Kanfer, G., Kolar, K., Lang, A.,
Michel, A. H., and Kornmann, B. (2013) Biochim. Biophys. Acta,
DOI: 10.1016/j.bbamcr.2013.01.028.
30.Rizzuto, R., Pinton, P., Carrington, W., Fay, F.
S., Fogarty, K. E., Lifshitz, L. M., Tuft, R. A., and Pozzan, T. (1998)
Science, 280, 1763-1766.
31.Csordas, G., Renken, C., Varnai, P., Walter, L.,
Weaver, D., Buttle, K. F., Balla, T., Mannella, C. A., and Hajnoczky,
G. (2006) J. Cell Biol., 174, 915-921.
32.Csordas, G., Varnai, P., Golenar, T., Roy, S.,
Purkins, G., Schneider, T. G., Balla, T., and Hajnoczky, G. (2010)
Mol. Cell, 39, 121-132.
33.Stern, J. E., and Filosa, J. A. (2013) Auton.
Neurosci., 175, 51-60.
34.Araque, A., Parpura, V., Sanzgiri, R. P., and
Haydon, P. G. (1999) Trends Neurosci., 22, 208-215.
35.Volterra, A., and Meldolesi, J. (2005) Nat.
Rev. Neurosci., 6, 626-640.
36.Zonta, M., Angulo, M. C., Gobbo, S., Rosengarten,
B., Hossmann, K. A., Pozzan, T., and Carmignoto, G. (2003) Nat.
Neurosci., 6, 43-50.
37.Rakic, P. (2003) Cereb. Cortex, 13,
541-549.
38.Giaume, C., Koulakoff, A., Roux, L., Holcman, D.,
and Rouach, N. (2010) Nat. Rev. Neurosci., 11, 87-99.
39.McClanahan, T., Nao, B., Wolke, L., Martin, B.,
Mertz, T., and Gallagher, K. A. (1993) FASEB J., 7,
A118.
40.Takaoka, A., Nakae, I., Mitsunami, K., Yabe, T.,
Morikawa, S., Inubushi, T., and Kinoshita, M. (1999) J. Am. Coll.
Cardiol., 33, 556-564.
41.Tapuria, N., Kumar, Y., Habib, M. M., Abu Amara,
M., Seifalian, A. M., and Davidson, B. R. (2008) J. Surg. Res.,
150, 304-330.
42.Silachev, D. N., Isaev, N. K., Pevzner, I. B.,
Zorova, L. D., Stelmashook, E. V., Novikova, S. V., Plotnikov, E. Y.,
Skulachev, V. P., and Zorov, D. B. (2012) PLoS One, 7,
e51553.
43.Ernster, L., Ikkos, D., and Luft, R. (1959)
Nature, 184, 1851-1854.
44.Luft, R., Ikkos, D., Palmieri, G., Ernster, L.,
and Afzelius, B. (1962) J. Clin. Invest., 41,
1776-1804.
45.Schaefer, A. M., McFarland, R., Blakely, E. L.,
He, L., Whittaker, R. G., Taylor, R. W., Chinnery, P. F., and Turnbull,
D. M. (2008) Ann. Neurol., 63, 35-39.
46.Elliott, H. R., Samuels, D. C., Eden, J. A.,
Relton, C. L., and Chinnery, P. F. (2008) Am. J. Hum. Genet.,
83, 254-260.
47.Manwaring, N., Jones, M. M., Wang, J. J.,
Rochtchina, E., Howard, C., Mitchell, P., and Sue, C. M. (2007)
Mitochondrion, 7, 230-233.
48.Gray, M. W., Burger, G., and Lang, B. F. (1999)
Science, 283, 1476-1481.
49.Nass, M. M., and Nass, S. (1963) J. Cell
Biol., 19, 593-611.
50.Holt, I. J., Harding, A. E., and Morgan-Hughes,
J. A. (1988) Nature, 331, 717-719.
51.Wallace, D. C., Singh, G., Lott, M. T., Hodge, J.
A., Schurr, T. G., Lezza, A. M., Elsas, L. J., 2nd, and Nikoskelainen,
E. K. (1988) Science, 242, 1427-1430.
52.Dimauro, S., and Rustin, P. (2009) Biochim.
Biophys. Acta, 1792, 1159-1167.
53.Greaves, L. C., Reeve, A. K., Taylor, R. W., and
Turnbull, D. M. (2012) J. Pathol., 226, 274-286.
54.Eleff, S., Kennaway, N. G., Buist, N. R.,
Darley-Usmar, V. M., Capaldi, R. A., Bank, W. J., and Chance, B. (1984)
Proc. Natl. Acad. Sci. USA, 81, 3529-3533.
55.Jokinen, R., and Battersby, B. J. (2012) Ann.
Med., 45, 149-155.
56.Rossignol, R., Faustin, B., Rocher, C., Malgat,
M., Mazat, J. P., and Letellier, T. (2003) Biochem. J.,
370, 751-762.
57.Lee, H. S., Ma, H., Juanes, R. C., Tachibana, M.,
Sparman, M., Woodward, J., Ramsey, C., Xu, J., Kang, E. J., Amato, P.,
Mair, G., Steinborn, R., and Mitalipov, S. (2012) Cell Rep.,
1, 506-515.
58.Taylor, R. W., Wardell, T. M., Smith, P. M.,
Muratovska, A., Murphy, M. P., Turnbull, D. M., and Lightowlers, R. N.
(2001) Adv. Drug. Deliv. Rev., 49, 121-125.
59.Dianov, G. L., Souza-Pinto, N., Nyaga, S. G.,
Thybo, T., Stevnsner, T., and Bohr, V. A. (2001) Prog. Nucleic Acid.
Res. Mol. Biol., 68, 285-297.
60.De Souza-Pinto, N. C., Mason, P. A., Hashiguchi,
K., Weissman, L., Tian, J., Guay, D., Lebel, M., Stevnsner, T. V.,
Rasmussen, L. J., and Bohr, V. A. (2009) DNA Repair (Amst.),
8, 704-719.
61.Vorobjev, I. A., and Zorov, D. B. (1983) FEBS
Lett., 163, 311-314.
62.Hermann, G. J., and Shaw, J. M. (1998) Annu.
Rev. Cell Dev. Biol., 14, 265-303.
63.Skulachev, V. P., Bakeeva, L. E., Chernyak, B.
V., Domnina, L. V., Minin, A. A., Pletjushkina, O. Y., Saprunova, V.
B., Skulachev, I. V., Tsyplenkova, V. G., Vasiliev, J. M., Yaguzhinsky,
L. S., and Zorov, D. B. (2004) Mol. Cell Biochem.,
256-257, 341-358.
64.Bakeeva, L. E., Chentsov, Yu. S., and Skulachev,
V. P. (1978) Biochim. Biophys. Acta, 501, 349-369.
65.Amchenkova, A. A., Bakeeva, L. E., Chentsov, Y.
S., Skulachev, V. P., and Zorov, D. B. (1988) J. Cell Biol.,
107, 481-495.
66.Poliakova, I. A., Zorov, D. B., and Leikina, M.
I. (1995) Dokl. Akad. Nauk, 342, 553-555.
67.Plotnikov, E. Y., Vasileva, A. K.,
Arkhangelskaya, A. A., Pevzner, I. B., Skulachev, V. P., and Zorov, D.
B. (2008) FEBS Lett., 582, 3117-3124.
68.Lightowlers, R. N., Chinnery, P. F., Turnbull, D.
M., and Howell, N. (1997) Trends Genet., 13, 450-455.
69.Khryapenkova, T. G., Plotnikov, E. Y.,
Korotetskaya, M. V., Sukhikh, G. T., and Zorov, D. B. (2008) Byul.
Eksp. Biol. Med., 146, 506-511.
70.De Duve, C., and Wattiaux, R. (1966) Annu.
Rev. Physiol., 28, 435-492.
71.Glaumann, H., Berezesky, I. K., Ericsson, J. L.,
and Trump, B. F. (1975) Lab. Invest., 33, 239-251.
72.Wassermann, V. A., Neisser, A., and Bruck, C.
(1906) Dtsch. Med. Wochenschr., 32, 745.
73.Doniach, D., and Walker, G. (1974) Gut,
15, 664-668.
74.Carafoli, E. (1986) Ann. N. Y. Acad. Sci.,
488, 1-18.
75.Baum, H. (1995) Biochim. Biophys. Acta,
1271, 111-121.
76.Krysko, D. V., Agostinis, P., Krysko, O., Garg,
A. D., Bachert, C., Lambrecht, B. N., and Vandenabeele, P. (2011)
Trends Immunol., 32, 157-164.
77.Shimada, K., Crother, T. R., Karlin, J.,
Dagvadorj, J., Chiba, N., Chen, S., Ramanujan, V. K., Wolf, A. J.,
Vergnes, L., Ojcius, D. M., Rentsendorj, A., Vargas, M., Guerrero, C.,
Wang, Y., Fitzgerald, K. A., Underhill, D. M., Town, T., and Arditi, M.
(2012) Immunity, 36, 401-414.
78.Zorov, D. B. (1996) Biochim. Biophys.
Acta, 1275, 10-15.
79.Tuppen, H. A., Blakely, E. L., Turnbull, D. M.,
and Taylor, R. W. (2010) Biochim. Biophys. Acta, 1797,
113-128.
80.Mitchell, P. (1961) Nature, 191,
144-148.
81.Mitchell, P. (1966) Biol. Rev. Camb. Philos.
Soc., 41, 445-502.
82.Liberman, E. A., Topaly, V. P., Tsofina, L. M.,
Jasaitis, A. A., and Skulachev, V. P. (1969) Nature, 222,
1076-1078.
83.Zorov, D. B., Krasnikov, B. F., Kuzminova, A. E.,
Vysokikh, M., and Zorova, L. D. (1997) Biosci. Rep., 17,
507-520.
84.Zorov, D. B., Isaev, N. K., Plotnikov, E. Y.,
Zorova, L. D., Stelmashook, E. V., Vasileva, A. K., Arkhangelskaya, A.
A., and Khrjapenkova, T. G. (2007) Biochemistry (Moscow),
72, 1115-1126.
85.Zamzami, N., Susin, S. A., Marchetti, P., Hirsch,
T., Gomez-Monterrey, I., Castedo, M., and Kroemer, G. (1996) J. Exp.
Med., 183, 1533-1544.
86.Liu, X., Kim, C. N., Yang, J., Jemmerson, R., and
Wang, X. (1996) Cell, 86, 147-157.
87.Hershko, A., Ciechanover, A., and Rose, I. A.
(1979) Proc. Natl. Acad. Sci. USA, 76, 3107-3110.
88.Ciechanover, A. (1994) Cell, 79,
13-21.
89.Sutovsky, P., Moreno, R. D., Ramalho-Santos, J.,
Dominko, T., Simerly, C., and Schatten, G. (1999) Nature,
402, 371-372.
90.Jin, S. M., Lazarou, M., Wang, C., Kane, L. A.,
Narendra, D. P., and Youle, R. J. (2010) J. Cell Biol.,
191, 933-942.
91.Di Lisa, F., Blank, P. S., Colonna, R., Gambassi,
G., Silverman, H. S., Stern, M. D., and Hansford, R. G. (1995) J.
Physiol., 486, 1-13.
92.Catia Sorgato, M., Lippe, G., Seren, S., and
Ferguson, S. J. (1985) FEBS Lett., 181, 323-327.
93.Skulachev, V. P. (1996) Q. Rev. Biophys.,
29, 169-202.
94.Korshunov, S. S., Skulachev, V. P., and Starkov,
A. A. (1997) FEBS Lett., 416, 15-18.
95.Starkov, A. A. (1997) Biosci. Rep.,
17, 273-279.
96.Cunha, F. M., Caldeira da Silva, C. C.,
Cerqueira, F. M., and Kowaltowski, A. J. (2011) Curr. Drug
Targets, 12, 783-789.
97.Plotnikov, E. Y., Silachev, D. N., Jankauskas, S.
S., Rokitskaya, T. I., Chupyrkina, A. A., Pevzner, I. B., Zorova, L.
D., Isaev, N. K., Antonenko, Y. N., Skulachev, V. P., and Zorov, D. B.
(2012) Biochemistry (Moscow), 77, 1029-1037.
98.Martineau, L. C. (2012) Biochim. Biophys.
Acta, 1820, 133-150.
99.Chen, K., Kobayashi, S., Xu, X., Viollet, B., and
Liang, Q. (2013) PLoS One, 8, e59682.
100.Izyumov, D. S., Avetisyan, A. V., Pletjushkina,
O. Y., Sakharov, D. V., Wirtz, K. W., Chernyak, B. V., and Skulachev,
V. P. (2004) Biochim. Biophys. Acta, 1658, 141-147.
101.Shabalina, I. G., and Nedergaard, J. (2011)
Biochem. Soc. Trans., 39, 1305-1309.
102.Zorov, D. B., Filburn, C. R., Klotz, L. O.,
Zweier, J. L., and Sollott, S. J. (2000) J. Exp. Med.,
192, 1001-1014.
103.Zorov, D. B., Juhaszova, M., and Sollott, S. J.
(2006) Biochim. Biophys. Acta, 1757, 509-517.
104.Aon, M. A., Cortassa, S., and O’Rourke,
B. (2008) Adv. Exp. Med. Biol., 641, 98-117.
105.O’Rourke, B., Ramza, B. M., and Marban,
E. (1994) Science, 265, 962-966.
106.O’Rourke, B. (2000) J. Physiol.,
529, 23-36.
107.Aon, M. A., Cortassa, S., Marban, E., and
O’Rourke, B. (2003) J. Biol. Chem., 278,
44735-44744.
108.Akar, F. G., Aon, M. A., Tomaselli, G. F., and
O’Rourke, B. (2005) J. Clin. Invest., 115,
3527-3535.
109.Aon, M. A., Cortassa, S., Akar, F. G., and
O’Rourke, B. (2006) Biochim. Biophys. Acta, 1762,
232-240.
110.Aon, M. A., Cortassa, S., Akar, F. G., Brown,
D. A., Zhou, L., and O’Rourke, B. (2009) Int. J. Biochem. Cell
Biol., 41, 1940-1948.
111.Zhou, L., Aon, M. A., Liu, T., and
O’Rourke, B. (2011) J. Mol. Cell Cardiol., 51,
632-639.
112.McEnery, M. W., Snowman, A. M., Trifiletti, R.
R., and Snyder, S. H. (1992) Proc. Natl. Acad. Sci. USA,
89, 3170-3174.
113.Kinnally, K. W., Zorov, D. B., Antonenko, Y.
N., Snyder, S. H., McEnery, M. W., and Tedeschi, H. (1993) Proc.
Natl. Acad. Sci. USA, 90, 1374-1378.
114.Skulachev, V. P., Anisimov, V. N., Antonenko,
Y. N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., Filenko, O. F.,
Kalinina, N. I., Kapelko, V. I., Kolosova, N. G., Kopnin, B. P.,
Korshunova, G. A., Lichinitser, M. R., Obukhova, L. A., Pasyukova, E.
G., Pisarenko, O. I., Roginsky, V. A., Ruuge, E. K., Senin, I. I.,
Severina, I. I., Skulachev, M. V., Spivak, I. M., Tashlitsky, V. N.,
Tkachuk, V. A., Vyssokikh, M. Y., Yaguzhinsky, L. S., and Zorov, D. B.
(2009) Biochim. Biophys. Acta, 1787, 437-461.
115.Omenn, G. S., Goodman, G. E., Thornquist, M.
D., Balmes, J., Cullen, M. R., Glass, A., Keogh, J. P., Meyskens, F.
L., Jr., Valanis, B., Williams, J. H., Jr., Barnhart, S., Cherniack, M.
G., Brodkin, C. A., and Hammar, S. (1996) J. Natl. Cancer Inst.,
88, 1550-1559.
116.Albanes, D., Heinonen, O. P., Taylor, P. R.,
Virtamo, J., Edwards, B. K., Rautalahti, M., Hartman, A. M., Palmgren,
J., Freedman, L. S., Haapakoski, J., Barrett, M. J., Pietinen, P.,
Malila, N., Tala, E., Liippo, K., Salomaa, E. R., Tangrea, J. A.,
Teppo, L., Askin, F. B., Taskinen, E., Erozan, Y., Greenwald, P., and
Huttunen, J. K. (1996) J. Natl. Cancer Inst., 88,
1560-1570.
117.Hercberg, S. (2005) Am. J. Clin. Nutr.,
81, 218S-222S.
118.Lichtenstein, A. H., Appel, L. J., Brands, M.,
Carnethon, M., Daniels, S., Franch, H. A., Franklin, B., Kris-Etherton,
P., Harris, W. S., Howard, B., Karanja, N., Lefevre, M., Rudel, L.,
Sacks, F., van Horn, L., Winston, M., and Wylie-Rosett, J. (2006)
Circulation, 114, 82-96.
119.Sesso, H. D., Buring, J. E., Christen, W. G.,
Kurth, T., Belanger, C., MacFadyen, J., Bubes, V., Manson, J. E.,
Glynn, R. J., and Gaziano, J. M. (2008) Jama, 300,
2123-2133.
120.Azzi, A. (2009) IUBMB Life, 61,
1159-1160.
121.Han, S. N., Pang, E., Zingg, J. M., Meydani, S.
N., Meydani, M., and Azzi, A. (2010) Arch. Biochem. Biophys.,
495, 49-55.
122.Zingg, J. M., and Azzi, A. (2004) Curr. Med.
Chem., 11, 1113-1133.
123.Lewis, K. N., Andziak, B., Yang, T., and
Buffenstein, R. (2013) Antioxid. Redox. Signal., DOI:
10.1089/ars.2012.4911.
124.Bakeeva, L. E., Barskov, I. V., Egorov, M. V.,
Isaev, N. K., Kapelko, V. I., Kazachenko, A. V., Kirpatovsky, V. I.,
Kozlovsky, S. V., Lakomkin, V. L., Levina, S. B., Pisarenko, O. I.,
Plotnikov, E. Y., Saprunova, V. B., Serebryakova, L. I., Skulachev, M.
V., Stelmashook, E. V., Studneva, I. M., Tskitishvili, O. V.,
Vasilyeva, A. K., Victorov, I. V., Zorov, D. B., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1288-1299.
125.Plotnikov, E. Y., Silachev, D. N., Chupyrkina,
A. A., Danshina, M. I., Jankauskas, S. S., Morosanova, M. A.,
Stelmashook, E. V., Vasileva, A. K., Goryacheva, E. S., Pirogov, Y. A.,
Isaev, N. K., and Zorov, D. B. (2010) Biochemistry (Moscow),
75, 145-150.
126.Skulachev, M. V., Antonenko, Y. N., Anisimov,
V. N., Chernyak, B. V., Cherepanov, D. A., Chistyakov, V. A., Egorov,
M. V., Kolosova, N. G., Korshunova, G. A., Lyamzaev, K. G., Plotnikov,
E. Y., Roginsky, V. A., Savchenko, A. Y., Severina, I. I., Severin, F.
F., Shkurat, T. P., Tashlitsky, V. N., Shidlovsky, K. M., Vyssokikh, M.
Y., Zamyatnin, A. A., Jr., Zorov, D. B., and Skulachev, V. P. (2012)
Curr. Drug Targets, 12, 800-826.
127.Plotnikov, E. Y., Chupyrkina, A. A.,
Jankauskas, S. S., Pevzner, I. B., Silachev, D. N., Skulachev, V. P.,
and Zorov, D. B. (2011) Biochim. Biophys. Acta, 1812,
77-86.
128.Ni Choileain, N., and Redmond, H. P. (2006)
Surgeon, 4, 23-31.
129.Lenz, A., Franklin, G. A., and Cheadle, W. G.
(2007) Injury, 38, 1336-1345.
130.Simmons, E. M., Himmelfarb, J., Sezer, M. T.,
Chertow, G. M., Mehta, R. L., Paganini, E. P., Soroko, S., Freedman,
S., Becker, K., Spratt, D., Shyr, Y., and Ikizler, T. A. (2004)
Kidney Int., 65, 1357-1365.