REVIEW: Plasticity of tumor cell Migration: acquisition of new properties or return to the Past?
A. Y. Alexandrova
Institute of Carcinogenesis, Blokhin Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, 115478 Moscow, Russia; E-mail: tonya_alex@yahoo.com
Received May 8, 2014; Revision received June 5, 2014
During tumor development cancer cells pass through several stages when cell morphology and migration abilities change remarkably. These stages are named epithelial–mesenchymal and mesenchymal–amoeboid transitions. The molecular mechanisms underlying cell motility are changing during these transitions. As result of transitions the cells acquire new characteristics and modes of motility. Cell migration becomes more independent from the environmental conditions, and thus cell dissemination becomes more aggressive, which leads to formation of distant metastases. In this review we discuss the characteristics of each of the transitions, cell morphology, and the specificity of cellular structures responsible for different modes of cell motility as well as molecular mechanisms regulating each transition.
KEY WORDS: epithelial–mesenchymal transition, mesenchymal–amoeboid transition, actin cortex, lamellipodia, filopodia, bleb, small GTPasesDOI: 10.1134/S0006297914090107
Abbreviations: AMT, amoeboid–mesenchymal transition; ECM, extracellular matrix; EMT, epithelial–mesenchymal transition; MAT, mesenchymal–amoeboid transition; MMPs, matrix metalloproteinases.
Tumor development is closely associated with the acquisition of the
ability of tumor cells for increased migration, which permits cells to
escape from the border of afflicted tissue and leads to the penetration
of cells into adjacent organs and tissues (invasion) and dissemination
of tumor cells to distant organs with subsequent formation of new tumor
nodules there (metastasis). These processes are the main causes of
death of oncological patients. Tumor cells need to show completely new
features to facilitate their effective migration under new conditions
that permit them to pass interstitial barriers, penetrate through the
basal membrane and different kinds of extracellular matrix (ECM), as
well as to intravasate into lymphatic and blood vessels for
distribution to remote areas of the body. It is believed that the
acquisition of new properties of tumor cells supporting their unlimited
proliferation and facilitating their dissemination and survival in new
environments is underlain by instability of their genome [1], which leads to the emergence and consolidation of
new adaptations and thus to tumor progression. Each of these properties
(enhanced migration activity, unlimited proliferation, and survival in
nonoptimal conditions) is achieved by complex molecular mechanisms. For
example, the acquisition of so-called motile phenotype by cells is
associated with remarkable alterations of their cytoskeleton,
disturbance of cell–cell contacts, changes in cell morphology,
increase in matrix metalloproteinase (MMP) activity, and with
alterations of many other characteristics. Of course the acquisition of
new properties could occur gradually and is explained by quantitative
changes in the production or activity of a particular protein. However,
there are several basic steps that are associated with the switch of
cells to a new mode of movement that is based on molecular mechanisms
that are different from those used previously. Such changes are
qualitative in nature and are called “transitions”. It is
possible to identify two such transitions.
The epithelial–mesenchymal transition (EMT) is when cells previously connected by cell–cell contacts to integrated epithelial sheet begin to lose these contacts and to move as individual cells by a motility mode like fibroblasts (mesenchymal cells) use.
The mesenchymal–amoeboid transition (MAT) is when cells that previously moved like fibroblasts by the mesenchymal mode of motility change their shape to more or less round and begin to move using very special type of protrusions – blebs, similar to motility of the amoeba Dictyostelium, thus transiting to the amoeboid mode of motility.
Each of these transitions gives cells certain advantages in moving, enhances migration effectiveness, and thus the effectiveness of cell dissemination. There are different molecular mechanisms as the basis of each motility mode, and thus transition means changeover of these mechanisms. The ability of cells to change the mechanisms of movement depending on external conditions and intracellular regulation is called plasticity. We will consider each mode of motility, the morphological characteristics of cells migrating by different modes, the cellular and molecular mechanisms underlying each type of motility, and different pathways regulating the transitions. Plasticity was shown for tumor cells, and it is adaptations that arise during tumor progression that facilitate invasion and metastasis. The question of at what stage of tumor development this property appears and whether it is a common biological property of the cells or an exclusive feature of cancer cells, in my opinion, is of particular interest and is important for understanding general mechanisms underlying motility of tumor cells.
EPITHELIAL–MESENCHYMAL TRANSITION
Epithelial–mesenchymal transition (EMT) is the complex reorganization that means transition from low motile epithelial cells of different organs integrated with each other into epithelial sheet to highly motile individual cells. Under normal conditions fibroblasts (mesenchymal cells) move as individuals, for example during wound healing, so such mode of migration is called mesenchymal. Transition of cells from collective to individual migration followed by loss of epithelial characteristics and acquisition of fibroblasts-like features is called EMT [2-4]. EMT occurs in certain situations, for example during wound healing, embryonic development [5, 6], and in tumor progression. The main features of EMT are disruption of cell–cell contacts, disorder of basal–apical cell polarity, actin cytoskeleton reorganization, and acquisition of “mobile” phenotype. EMT is an intensively studied process. It is followed by significant modifications in transcription of different genes [7, 8]. These modifications are regulated by transcriptional factors such as Snail, Twist, Slug, ZEB1, ZEB2, Lef-1, b-catenin, etc. EMT leads to appearance of polarized cells with pronounced front and back part [9]. Cell migration is provided by protrusions on the front (leading) part and by contraction of the back (tail) part. The typical feature of mesenchymal migration is secretion by cells of proteolytic enzymes, matrix metalloproteinases (MMPs) and urokinase-type plasminogen activator (uPA), which are responsible for degradation of extracellular matrix (ECM) [10-12]. Due to degradation of ECM, tumor cells make a path to go through tissues and overcome tissue barriers. The main protrusions formed during mesenchymal migration and which are morphological features of this type of motility are lamellipodia (a plate at the leading end outgrowth cells) and filopodia (narrow cylindrical outgrowths). The actin cytoskeleton of cells using the mesenchymal mode of migration consists mainly of bundles of actin filaments – stress fibers. Below we will consider in more details the organization and mechanisms of formation of different protrusions responsible for cell migration.
MESENCHYMAL–AMOEBOID TRANSITION
This stage represents a change in motility patterns of individual cells and is based on a fundamental change in the molecular mechanisms governing the extension of protrusions on the leading edge of the cell. An external manifestation of mesenchymal–amoeboid transition (MAT) is dramatic changes in cell morphology, structure of the cytoskeleton, and the structure and dynamics of protrusions, although cells continue to move individually (figure). The cells become rounded, actin stress fibers disappear, and the cytoskeleton organizes as actin cortex underlying the cell membrane. Cell motility by the amoeboid mode occurs due to formation very specialized protrusions – blebs [13, 14]. Cells using the amoeboid mode of motility form practically no focal adhesions with ECM, so this type of motility is independent of integrins [10, 15], they do not produce MMPs, and thereafter proteolytic degradation of ECM is absent [11, 16]. Unlike the EMT, the MAT is based not on deep changes in activity of transcriptional factors, but on quick phenotypic adaptations for a changing environmental conditions [17]. Thus, first, this transition can occur rather quickly, and second, the inverse transition from amoeboid to mesenchymal movement could easily occur in case of a reversal of the change in the environmental conditions. Amoeboid movements have been described for 3D substratum, possible because cells with absence of pronounced focal adhesions cannot properly attach to a 2D substratum, and in such experimental conditions all cells after MAT just detach from the substratum and go into the liquid medium. When comparing the rate of migration for cells using the mesenchymal and amoeboid motility modes, it is commonly noted that amoeboid motility is much faster than mesenchymal motility [16]. But to prove this thesis authors gave examples of quite different cells migrating using either amoeboid or mesenchymal mode. For example, Friedl and coauthors showed that the rate of a cell using the mesenchymal mode of migration on a 3D substratum (melanoma cells MV3 and fibroblasts) was 0.1-1 µm/min [18], while the rate of cells using amoeboid mode was from 2 µm/min (other melanoma cells A375m2) [19] and up to 25 µm/min (T-lymphocytes in collagen gel) [20]. Significant differences were shown in cell motion rate in 3D collagen matrix when the motilities of a few tumor cell lines (mesenchymal motility) and lymphocytes (amoeboid motility) were compared [21]. In one of the first works where MAT was demonstrated [11], the cells, which moved mesenchymally (fibrosarcoma cell line HT1080 and cells of breast carcinoma MDA-MB-231), were treated with proteinase inhibitor cocktail, and the authors expected a decline in cell movement due to cessation of matrix degradation. In spite of this expectation, such treatment did not cause the inhibition of motility, and the cells continued their migration in 3D even without proteolysis, which means that cells used mechanisms that did not require matrix degradation and thus should be other than mesenchymal. But the rate of the cells did not increase and was the same even after MAT. It was shown also that transition to amoeboid motility of prechordal plate progenitor cells during gastrulation of zebra fish (Danio rerio) induced by alteration of membrane-to-cortex attachment because of deficiency of proteins of the ERM family (ezrin, radixin, moesin) or activation of myosin II lead to reduction in rate and directionality of cell migration in comparison with normal prechordal plate progenitor cells moving by the mesenchymal mode with lamellipodia and filopodia [22]. Thus, it turns out that MAT is not likely needed to increase the rate of cell migration, but rather for migration of cells under changing environmental conditions, when, for one reason or another, the opportunity to use mesenchymal mode of motility declines (e.g. on inhibition of MMPs).
Actin cytoskeleton organization in protrusions of different types that provide for cell migration. a) Human skin fibroblast forms lamellipodia (Lp). b) MRC5 human lung fibroblasts transformed with virus SV40 (MRC5V2) form numerous filopodia (Fp) (photo by M. E. Lomakina, presented with kind permission). c) Stromal mesenchymal cell migrates using amoeboid mode due to blebs (B) formation (photo by A. S. Chikina, presented with kind permission). Upper row, the DIC microscope view of cells; middle row, schemes of actin cytoskeleton organization; the bottom row presents the main proteins involved in formation of the structures
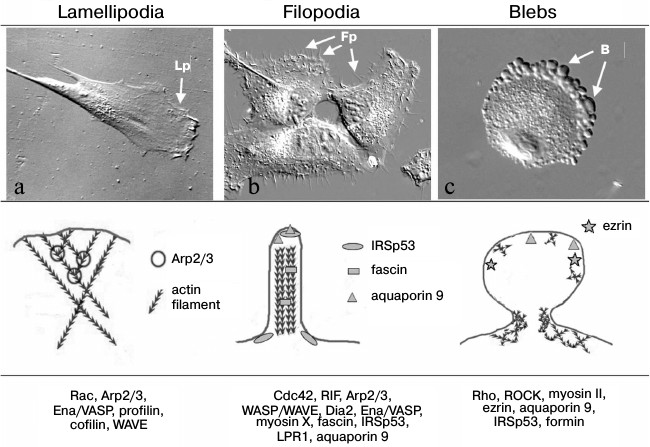
The migration of individual cells is usually followed by polarization and separation the leading edge with specialized protrusion structures and back tail part of the cell, which could pull up as the cell moves because of actin–myosin contractility, which lead to relocation of the cell. Polarization could be established due to external influences, for example chemotaxis. In the case when cells move mesenchymally in the direction of a chemotactic gradient, there are local increases in concentration of small GTPases Rac and Ras at the leading cell edge as a result of interaction of cellular receptors with chemotactic agents, activation of phosphatidylinositol-3-kinase (PI3K), and establishment of a gradient of phosphatidyl inositols [23-27]. But even without chemotaxis the polarity of mesenchymally migrating cells is established due to intracellular mechanisms. In particular, polarity could be determined by the local distribution and temporal activity of small GTPases of the Rho-family, which are important regulators of cytoskeletal reorganization. It has been shown that the small GTPases Rac1 and Cdc42 are activated at the leading edge of migrating cells [28-30]. The activation of small GTPases provided by local concentration of phosphatidylinositol(3,4,5)triphosphate (PIP3) on the leading cell edge [31] in turn leads to activation of guanine exchange factors (GEFs) that regulate activity of Rac and Cdc42 [32]. The active Rac stimulated Arp2/3-dependent polymerization of actin filaments and thus lamellipodial protrusion [33, 34]. Another small GTPase RhoA is activated in the tail region of cells, which leads to increase in actomyosin contractility and induced pull out of the tail region and reloading of the cell body [35-37]. The inhibition of local activity of Rac1, Cdc42, and RhoA lead to impairment of directional migration of neutrophils [38]. Also, a defect in local distribution of Rac1, Cdc42, and RhoA leads to a defect in formation of focal adhesions and in cell motility [39-41]. It was shown that even rather small changes in Rac activity lead to change in motility characteristics of moving fibroblasts. Increasing Rac activity promotes random or explorative cell migration, and vice versa decreasing Rac activity suppressed peripheral lamellae and switched the cell migration patterns of fibroblasts and epithelial cells from random to directionally persistent [42]. In a recent work using genetically encoded photoactivatable derivatives of Rac, it was shown that mutual regulation between Rac and Rho could be precisely spatial and temporarily local and is sufficient to determine polarized cell movement [43].
The other cytoskeleton system – microtubules – plays significant role in polarization of cells and in maintenance of directional cell movement [44]. Role of microtubules in organization of directional cell migration and mechanisms of their activity is discussed in details in excellent reviews [45, 46].
There are a few types of cell structures that provide protrusion of the leading edge (figure). The most common protrusions typical for different cell types are lamellipodia (broad, flat protrusion) and filopodia – narrow cylindrical protrusion, as well as more specific blebs (round membrane structure), invadopodia, podosomes [47], etc. These types of protrusions differ primarily in the structure of their actin cytoskeleton, as well as the dynamics and the molecular mechanisms governing their formation and pulling. Because protrusions are the main cell structures providing cell migration and their characteristics determine the effectiveness and character of migration, we will discuss below some of them in more detail.
Main structures localized on the leading cell edge and providing cell locomotion. As noted above, the most common protrusions by which the cell begins to move and push forward are lamellipodia and filopodia. The number of lamellipodia and filopodia is different in different cells. There are cells preferably extended through lamellipodia (fibroblasts) or predominantly due to filopodia (dendritic cells and neurons), and in some cells both structures may be represented equally (for example, B16 melanoma cells [48]).
Filopodia are cylindrical structures that can grow out to a few tens of microns from the cortical layer (figure) [49, 50]. Filopodia consist of 15-20 parallel actin filaments oriented by their (+)-ends toward the filopodial tip and packed into tight bundles [51-53]. Actin filaments are connected with each other via actin-associated proteins (for example, fimbrin or fascin) [49, 54-56]. Filopodia can be formed by cell of very different origin, and thus they can be very different in shape, molecular composition, and functionality (for reviews see [57, 58]). This review is devoted to cell movement, so we will mainly discuss filopodia on the leading cell edge that play role in cell motility and migration. Filopodia formation depends on activity of small GTPase Cdc42 [59]. It is assumed that during cell movement, filopodia have an explorative function. In particular, a cell can sense the gradient of a chemoattractant mainly due to filopodia, and this can determine the direction of cell migration [60-63]. Filopodia can also attach to a substratum through focal adhesions and thus a play role directly in reloading of cells [64]. A large set of proteins that regulate the actin cytoskeleton architecture has been shown to regulate filopodia formation. First of all it is proteins of the Ena/VASP family that promote formation of filopodia by preventing capping of plus ends of actin filaments [65, 66]. It was shown that among others the motor protein myosin X is quite important for formation of filopodia to transport other filopodial components, such as ENA/VASP proteins and integrins, to the dense tip of the filopodia [67, 68]; as well as protein IRSp53, which initiates filopodia by bending the membrane at the site of filopodia initiation through its inverse membrane-binding domain BAR (I-BAR) [69, 70], fascin which cross-links single parallel actin filaments into bundle inside filopodia [55], and the actin nucleator formin mDia2 which promotes nucleation of long unbranched filaments [71, 72]. The mechanisms of induction and formation of filopodia are discussed in detail in reviews by Mattila and Lappalainen [57] and Yang and Svitkina [58].
The lamellipodium is one of the main structures providing directed cell movement by the mesenchymal mode. It is thin flattened structure on the leading cell edge (figure). It has about 0.2 µm thickness and its width can be from 2 to 5 µm in dependence on cell type. The cytoskeleton of a lamellipodium consists of a thick uniform network of actin filaments. Using the method of decoration of actin filaments by fragments of heavy myosin chains, it was shown that all actin filaments at the leading edge have the same orientation, and their plus ends are directed to the plasma membrane [73]. When incorporation of fluorescein-fused G-actin into a filament was studied, it was found that a few minutes after injection the staining appeared at first at the periphery of the lamellipodium near the plasma membrane, then in small adhesion sites at the leading edge, and only later the staining could be seen in bundles and others actin structures [74, 75]. This means that polymerization of actin cytoskeleton occurred in lamellipodia and formation of other actin structures going rather through reorganization of the lamellipodial network than through polymerization of filaments de novo. In the process of polymerization, each actin filament produces force of a few piconewtons, which applied to membrane pushes it out [76]. It is believed that the main forces providing the leading cell edge protrusions produce in lamellipodia are due to actin polymerization [34, 53]. The molecular mechanism that underlies the operation and dynamics of the actin cytoskeleton in lamellipodia is Arp2/3-dependent actin polymerization [34, 77, 78]. The activators of Arp2/3 complex are proteins of the WASP family (Wiskott–Aldrich syndrome proteins). WASP provide the link between Cdc42- and Rac-dependent pathways and lead to lamellipodium and filopodium formation. Particularly one member of the WASP family – WAVE – localizes in lamellipodia. The rate of polymerization of new actin filaments depends on the number of globular actin monomers (G-actin). The protein cofilin severs and depolymerizes older actin filaments in the base of lamellipodia and thus lead to the renewal of the G-actin pool and stimulates branched network polymerization [78-80]. Profilin catalyzes exchange of ADP to ATP connected with G-actin and thus activates actin monomers and prepares them for future polymerization. Cell adhesion to substrate provided by special structures, focal adhesions [81-83], besides the mechanical cell–substrate connection are responsible for numerous regulatory functions [81, 84, 85]. It is believed that release of MMPs occurs in association with focal adhesions. Thus in experiments with HeLa cells and fibrosarcoma cells HT1080, it was shown by Takino et al. [86] that expression of membrane type 1 matrix metalloproteinase (MT1-MMP) as well as activation of MMP2 occur in areas of focal adhesions and lead to fibronectin (the ECM protein) disappearance from these regions. The biochemical evidence for matrix degradation were not presented in this work. The formation of nascent focal adhesions occurs mainly underneath lamellipodia, leads to attachment of lamellipodia to the substratum and thus determines its position [87, 88]. If a lamellipodium is not fixed on the substrate, it contracts with formation of riffles, which are the folds on the dorsal cell surface with dense actin network inside. The formation of nascent focal adhesions in the area of lamellipodia is the first step of actin cytoskeleton reorganization and leads to formation of bundles consisting of actin filament with opposite orientation and including myosin II molecules. These bundles are contractile and provide the relocation of the cell body during migration.
Podosomes and invadopodia are specialized structures providing both matrix degradation and cell invasion. Podosomes and invadopodia combine the characteristics of adhesion and protrusive structures. They are not as common as lamellipodia and filopodia; rather they are typical for specialized cells and are used mainly for migration of these cells in vivo. Podosomes are typical for osteoclasts, macrophages, some endothelial cells, and lymphocytes. Formation of podosomes is also described for many transformed cells of epithelial origin including HeLa and MCF-7 [89]. Podosomes are found on the ventral side of cells, and both provide cell adhesion and degradation of cell matrix with MMPs or serine proteases. Particularly lymphocytes ultimately form trans-cellular pores through the vascular endothelium for migration due to podosomes [90]. The podosomes are complex structures and include actin and cortactin regulating actin polymerization, as well as proteins typical for focal adhesions – vinculin and paxillin. Invasive cancer cells and cells transformed by oncogene src display podosome-like actin-rich membrane protrusions called invadopodia [91-93]. In both cases actin polymerization occurs by an Arp2/3-dependent mechanism. Sometimes podosomes and invadopodia are combined under the name invadosomes [94]. The differences between these two structures are rather small. These structures could be distinguished in size, dynamics, and their number per cell. Podosomes are as small as about 1 µm in diameter and 0.4 µm high, while invadopodia can reach 8 µm in diameter and 5 µm in height. There can be more than 20 podosomes but less than 10 (usually 1-2) invadopodia in one cell. Invadopodia are more stable, and their time of life can be as long as a few hours, while the lifetime of podosomes is in range of minutes [75, 93, 95]. Thereafter the level of matrix degradation is quite different in cases of podosomes and invadopodia in spite of both structures releasing MMP-2, MMP-9, and MT1-MMP [96-98]. Motility of cells with formation of podosomes and invadopodia is considered as mesenchymal migration.
Blebs are specialized rounded membrane protrusions (figure). For a long time it was believed that blebbing (bleb formation) is a morphological feature of apoptosis [99, 100]. During recent decades many data were collected showing that bleb formation can be the basis for a very special type of cell motility [13, 14]. The absence of apoptotic markers and nuclear fragmentation in cells forming blebs in the process of migration proves that such blebbing is not associated with apoptosis. Cells which move due to bleb formation are similar to moving amoebae Dictyostelium discoideum and Entamoeba histolytica in shape (rounded) and according types of protrusions (blebs), and thereafter such type of motility was named amoeboid [101, 102]. It was shown that blebbing occurs due to increase in intracellular pressure, for example, as a result of contraction of actomyosin after activation of small GTPase Rho or myosin II [103]. At the beginning of formation, blebs are just membrane protrusions and there is no underlying actin cytoskeleton inside them [104-109]. The initiation of blebbing could be a result either of disruption of connection between cell membrane and submembrane actin cortex or local defect or depolymerization of actin cortex itself [13, 14, 107]. After membrane protrusion, the formation of actin cytoskeleton begins under the expanding membrane. Polymerization of actin cortex during blebbing is regulated by formins, which are nucleotide polymerization factors different from those that participate in polymerization of branched actin network in lamellipodia [110-112]. Then the contraction of this newly formed actin cortex leads to retraction of the blebs [107]. The relocation of blebbing cells happens because blebs expand into pores of surrounded matrix and become mechanically fixed in these pores, then due to actomyosin contraction the body of the cell is pulled toward the fixed bleb. Mechanisms of motility driven by blebbing are discussed in detail in a review by Paluch and Raz [113].
Blebs could form both on the whole cell surface or polarized to one cell edge to make this edge leading and thus determining the direction of motility. Such polarized blebbing could be induced by interactions with chemokines. For example, polarized bleb formation and directed migration of cells driven by blebbing was described for embryogenesis under the action of SDF1 (stromal cell-derived factor 1), which is a chemokine of subfamily CXC playing an important role in embryonic development and hemopoiesis. Besides that, polarity of blebbing could be provided by local differences in intracellular distribution of some proteins. Bleb formation depends on interaction between plasma membrane and actin cortex and thus activity and distribution of proteins regulating these interactions are quite important for regulation of blebbing. An example of such proteins is members of the ERM family – ezrin, radixin, and moesin. Ezrin increases the link between plasma membrane and cytoskeleton. It was shown that activation of ezrin, radixin, and moesin correlated with decline of blebbing during motility of germ cells of Danio rerio [114], melanoma cells A375 [115], and mast cells [116]. It was also shown that in motile Walker carcinosarcoma cells the ezrin concentration is increased in the tail part of cells, which leads to activation of membrane–cortex coupling, and conversely is decreased at the leading edge, which causes weakening of membrane–cortex link and promotes the polarized formation of blebs [117-119].
It was also shown that even non-polarized blebbing leads to increase in invasive capacities of cells [110, 120, 121]. This fact gives evidence that amoeboid movement, or the cell’s ability to change the mechanism of movement under certain conditions, provides them with an additional advantage in dissemination.
The important question of the compatibility of results of cell migration obtained in studies in vitro and under in vivo conditions still remains open. Amoeboid movement was mainly studied in 3D matrixes. Cells moving by blebbing do not form focal adhesions with ECM, and thus they do not release MMPs during motility. Sabeh et al. [122] showed that 3D matrixes used in several studies are quite different from natural tumor ECM and thus there are different requirements for MMPs under in vitro and in vivo conditions. According to their data, the activity of at least one metalloproteinase, particularly the membrane-type 1 matrix metalloproteinase (MT1-MMP), is necessary for cell migration in vivo, and without it the invasion of breast carcinoma cells was not observed. According to these data, migration independent of matrix degradation could occur only in the case when the collagen matrix is devoid of transverse connections, which are usually present in tissues.
Factors regulating plasticity of migration. Plasticity of migration is the ability of cells to switch between different modes of motility, and it allows cells to “choose” the mechanism of migration in accordance with the structure of the surrounding ECM. What factors determine the existence of such ability?
As mentioned above, the main regulators of cytoskeleton reorganization that determine the character of cell motility are small GTPases of the Rho family [59, 123]. It was shown that for cells using mesenchymal mode of motility with formation of lamellipodia and filopodia the most important are Rac and Cdc42 GTPases, respectively. In contrast, for amoeboid motility the small GTPase Rho is the most significant. It activates the kinases ROCK I and II, which leads to increase in contractility of underlying membrane actin cortex [11, 124, 125]. Experiments with regulation of GTPase activity using small interfering RNA (siRNA) to guanine nucleotide exchange factors (GEFs) and GTPase accelerating proteins (GAPs) show that it is the balance of small GTPases that determines plasticity of mechanisms of cell movement [112]. Thus, increased activity of Rac leads to the formation of lamellipodia and suppresses amoeboid movement, and conversely during amoeboid movement Rac activity is suppressed. It was shown also in other works that inhibition of Rac activity due to overexpression of FilGAP lead to increase in Rho activity and thus stimulation of blebbing [126]. However, not even all cancer cells are capable of demonstrating plasticity under the same conditions. In experiments with glioblastoma cells U87MG, which moved in 3D substrate by the mesenchymal mode, Rac inhibition stopped migration, while in the case of fibrosarcoma cells HT1080 the same influence lead to MAT. At the same time, the inhibition of both Rho and Rac in these cells leads to complete halt of migration [127]. Hereby the balance of small GTPases is a subtle and sensitive mechanism determining the migratory behavior of cells. It is not yet clear what outside and inside factors regulate these relationships.
Transition to amoeboid movement could happen as a result of very different influences limiting or disturbing mesenchymal movement. It could be disturbance in substrate adhesiveness [128] as well as alteration of its density or architecture [129, 130]. Cells MDA-MB-435 demonstrated mesenchymal movement on plastic dishes, but changed to rounded shape and blebbing on or inside Matrigel (ECM consisting from basal membrane proteins) [110].
Another approach stopping mesenchymal movement of cells in one case and to switching MAT in other case is inhibition of Arp2/3-dependent actin polymerization with specialized inhibitor CK666. Treatment of HT1080 cells with CK666 caused the MAT (our unpublished data). In the case of migration of glioma cells (lines U251, LN229, and SNB19) on two-dimensional substrate, the inhibition of Arp2/3 with CK666 only inhibited their migration [131].
One of the influences limiting mesenchymal motility is treatment of cells with MMPs inhibitors, which was the condition for the first demonstration of MAT [11, 132]. The molecular mechanisms that could explain why in some cases the limitation of mesenchymal motility leads to MAT while in others not are absolutely unclear. For now the most likely explanation is the total increase in small GTPases activity in tumor cells when inhibition or limitation of one mechanism leads to stimulation of another on the basis of the balance of GTPases.
The mesenchymal and amoeboid modes of movement are not entirely mutually exclusive. During early embryonic development simultaneous formation of lamellipodia and blebs by the same cells were demonstrated [22]. In our experiments the inhibition of Arp2/3 activity with CK666 in tumor cells leads first to significant increase the number of filopodia and then to blebbing. Similar preliminary formation of numerous filopodia and only later appearance of blebs was shown when mesenchymal migration of tumor cells was limited due to the deterioration of substrate adhesiveness after coating it with PolyHema (poly(2-hydroxyethyl methacrylate)) solutions of different concentrations (A. S. Chikina, A. Y. Alexandrova, unpublished). We could see that although filopodia are typical protrusions for the mesenchymal mode of motility, a stage with numerous filopodia could be transitory between lamellipodia (Arp2/3-dependent actin polymerization, mesenchymal motility) and blebs (formin-dependent actin polymerization, amoeboid motility) in the process of MAT when the molecular mechanisms regulated motility change. To explain this phenomenon, we tried to analyze the common features between filopodia and blebs. Using cell precursors of mesendoderma in development of zebra fish embryos, it was shown that one of the mechanisms determining the type of protrusions could be regulation of link between actin cortex and plasma membrane [22]. One of cell mechanisms regulating relationships of membrane and underlying membrane actin cortex is the alteration of activity of aquaporins. Aquaporins belong to a family of intrinsic membrane proteins that act as selective channels for water, which due to rapid bidirectional flux of water can regulate local intracellular pressure [133]. The main function of aquaporins was postulated to be the regulation of water exchange in a wide range of organisms from plants to animals. The discovery of aquaporins resulted in the Nobel Prize in Chemistry for Peter Agre in 2003 together with Rodrick MacKinnon, who studied structure and mechanisms of action of potassium channels. Recently data were obtained showing that it is quite possible that aquaporins are involved in the regulation of cell motility. Particularly, it was shown that aquaporin 9 is localized on the leading edge of migrating neutrophils [134-138] and epithelial cells [139], and data showed that aquaporins can influence cell motility and angiogenesis [136, 137] and they are localized in special regions of plasma membrane associated with shape alterations and protrusion formation [140]. It was also shown that expression of aquaporin 9 induced filopodia formation in fibroblasts [141]. In further study of the role of aquaporins in cell motility, it was found that accumulation of aquaporin 9 in certain places in the plasma membrane induced formation of filopodia and blebs exactly at these points. Through the pores due to aquaporin 9, local water flux into the cell and at this point hydrostatic pressure is locally increased that leads to bleb formation [142]. In studying of the mechanisms of participation of aquaporins in initiation and formation of filopodia, it was found that growing filopodia have space free from actin filaments between the plasma membrane and cytoskeleton, and aquaporin 9 is concentrated at the tip of filopodia. The increased local water pressure in this point stimulates outgrowing of filopodia. It is interesting that aquaporins concentrated at the filopodial tip only at the moment of growing and quickly disappeared when growth stopped. Protrusion of filopodia occurs through transient formation of empty space at the filopodial tips, followed by actin assembly into this space to stabilize the protrusion. Thus the actin polymerization is delayed slightly compared to protrusion of filopodia, and actin cytoskeleton is required rather for long-term maintenance of filopodia than for elongation [142]. With these experiments the significant role of aquaporins for formation and elongation of filopodia was proved. In other work, it was demonstrated for liver endothelial cells that overexpression of aquaporin 1 leads to membrane blebbing as a response to FGF stimulation and thus stimulates liver endothelial cell invasion and angiogenesis during development of cirrhosis [143]. The connection between aquaporins and bleb formation is also discussed in other works [144]. Thus the participation of aquaporins in the formation of filopodia and blebs is proven. Concluding the analysis of these data, we can say that decrease in the link between plasma membrane and underlying membrane cytoskeleton is important for formation of both protrusion types. Whether aquaporins play a significant role in establishment of plasticity is yet unknown. There are only a few data about relations between aquaporins and tumor development. The elevation of aquaporin 4 expression was shown for glial tumors [145]. Probably the study of the role of aquaporins in the regulation of plasticity will be the subject of research in the near future.
A similar mechanism of formation of filopodia was shown by other authors [70]. This mechanism is based not on pushing the membrane due to polymerization of actin filaments, but on preliminary local protrusion of the plasma membrane caused by membrane deformation through activity of IRSp53. In this work the authors also observed preliminary development of membrane protrusion, and then actin filaments filled this protrusion. Thus actin polymerization was significantly more likely for stabilization of growing filopodia than for elongation itself [70]. In experiments with human embryonic kidney cells (HEK-293), it was shown that aquaporin 9 colocalized in filopodia with other filopodial proteins – myosin X and IRSp53 [142]. We did not find any data about the connection of IRSp53 with blebbing.
Thus the mechanisms underlying the growth of both filopodia and blebs require the disturbance of close physical interaction between cytoskeleton and plasma membrane and therefore are not so different. Perhaps that is why the stage with numerous filopodia appears as an intermediate in the transition from mesenchymal to amoeboid movement.
It should be noted that the important role of filopodia in directed cell migration has been known for a long time. It is widely known that tumor cells are characterized by the formation of numerous filopodia. Even more, there is direct evidence that increased expression of fascin 1, which is necessary for formation of filopodia, leads to development of migratory phenotype of prostate cancer cells and significantly increases the number of lung metastasis in in vivo experiments with mice [55]. Taking these data into account, we think that increased number of filopodia typical for cancer cells could be considered as the very beginning stage of MAT and therefore an indication that these cells may demonstrate plasticity of migratory mechanisms under changing external conditions.
Other players regulating connection of actin cytoskeleton and membrane are proteins of the ERM family (ezrin, radixin, moesin). These proteins both directly provide the link between actin cortex and plasma membrane through their ability to interact with transmembrane proteins and the underlying cytoskeleton and also can regulate the activities of signal transduction pathways responsible for cell polarity and migration [146]. It was shown that moesin participates in formation of membrane underlying actin cortex in non-polarized rounded lymphocytes and is accumulated in their tail region during migration (lymphocytes move using amoeboid motion) [147]. Ezrin promotes protrusion formation on the leading edge due to activation of Cdc42 [148]. It was hypothesized that different ERM proteins play different roles. Thus moesin stabilizes actin cortex in rounded cells and in the tail region of moving polarized cells and thus inhibit bleb formation. In contrast, ezrin impairs the link between cortex and membrane and promotes development of protrusions such as blebs, filopodia, and lamellipodia at the leading edge [149]. In some works it was shown that elevation of ERM proteins is associated with metastatic potential of tumor cells, the overexpression of ezrin being observed in metastasis during development of sarcoma [150, 151]. Expression of moesin increases remarkably in metastasis breast cancers, and the increase in moesin expression is associated with poor prognosis [152-154]. According the other data moesin depletion (which theoretically leads to depletion of the link between membrane and actin cytoskeleton and thus could promote MAT) increased the ability of melanoma cells to invade 3D collagen gel and to form lung metastasis in mice when injected into their tail vein [149]. In other work it was shown that ezrin depletion increases invasiveness to 3D matrix by colon cancer cells [155]. One could see that there are many different and sometimes opposite data about the role of ERM proteins in regulation of cell migration during tumor progression and about their possible role in regulation of transitions such as EMT and MAT [156]. Clarification of possible mechanisms of these activities of ERM proteins needs future investigation.
It is important to note that we should not consider the described transitions as constantly directed evolution of cell migration. At first both EMT and MAT could occur simultaneously in different cell populations of the same tumor. Also, these transitions are reversible, and the cells that went through MAT under certain conditions could switch back to mesenchymal movement. This ability to respond to external and internal conditions and adapt a migration mechanism to new conditions is apparently a property of tumor cells permitting and supporting their dissemination.
Sometimes the same factors could lead to either EMT or MAT in dependence on cellular context. In particular, it was shown that interaction with C-Met receptors followed by activation of tyrosine kinase Met could cause in some cases EMT and promotion of mesenchymal motility (for example scattering cells from epithelial islands under HGF/SF treatment [157]) and to transition to amoeboid motility and blebbing as happens in experiments with high metastatic potential breast cancer cells and with HEK293T cells (embryonic cells of human kidney transformed with SV-40 large T-antigen) (experimental activation of Met expression) [158].
CAN NORMAL CELLS DEMONSTRATE PLASTICITY OF MIGRATION
MECHANISMS?
As one can see from data noted above, the plasticity of cell migration mechanisms is mainly described for either cancer or embryonic cells. The question is – could normal differentiated cells demonstrate plasticity and go through transitions? Let us look at the different transitions.
EMT. EMT is observed in embryonic development and tumor progression and is regulated by switching of some transcriptional factors (see above). In contrast to MAT, it is rather easy to get experimental model of EMT because cells during this transition lose cell–cell contacts but still keep well pronounced focal adhesions with the substratum. Under experimental conditions EMT can be stimulated by different treatments, for example by activation of numerous signal transduction pathways, in particularly TGFβ, FGF, PDGF, Wnt, EGF, Ras-MAPK, Ras/PI3K/AKT, Hedgehog-GLI1, Notch, and others regulatory cascades [6-8], by treatment with cytokine HGF/SF [157, 159], Ras-transformation [160], and treatment of cells with the tumor promoter phorbol-12-myristate-13-acetate (PMA). The classical model of EMT with HGF/SF on MDCK cells is shown on specially selected clone of MDCK cells, called clone 20, and it does not work as well on non-selected cells [157]. PMA treatment causes EMT in ARCaP(E) prostate cancer cells [161]. In all other cases stimulation of EMT by different growth factors is usually caused by a significant surplus of activator compared to physiological conditions or by overexpression of regulatory factors (for example, HIF1 or NF-κB) [4]. Thereafter all classical models of EMT under experimental conditions are provided either with tumor cells or with specially selected cells, while differentiated epithelial cells demonstrate EMT only after extra strong treatment.
MAT. MAT is observed for tumor cell, usually for those that are on an advanced stage of tumor progression, as well for embryonic stem cells. It was shown that even not all tumor cells can go through MAT. For example, in migratory experiments with cells of breast carcinoma cells MBA-MB-468, prostate cancer cell line PC-3, and cells of colon carcinoma SW 480 in 3D collagen gel under treatment with MMP inhibitors, neither EMT no MAT was demonstrated [162]. These cells also did not demonstrate MAT in response to other approaches to slow mesenchymal motility, for example by inhibition of Rac or Arp2/3 activity, while other cells demonstrated MAT under the same conditions, for example, fibrosarcoma cells HT1080. According to our unpublished data, the mono oncogenic Ras-transformation did not stimulate the ability of fibroblasts to plasticity under the same conditions that lead to bleb formation by strongly transformed cells. There was only one work where we found that MAT was demonstrated on pseudo-normal cells of CHO line, when blebbing developed due to activation of membrane domain SH4 by Src-kinases [121], which promoted migration of stimulated cells into 3D matrix. It should be noted that src is a protooncogene and elevation of its expression leads to cell transformation. Interestingly, with other cells Src promotes formation of invadopodia and podosomes – cell protrusions that also promote invasion but by the mesenchymal mechanism of motility [163-165]. On the model of development of pancreatitis after stimulation of pancreatic acinus, the activation of Scr kinase also leads to blebbing associated with reorganization of actin cytoskeleton and cortactin redistribution, but these blebs were considered by the authors as signs of cell damage and not being involved in migration [166].
Leucocytes are cells that normally migrate using amoeboid mode of motility [167]. During inflammatory processes or infections, they migrate from the blood stream to the place of injury through tissue barriers and can demonstrate mutual MAT and AMT. It was shown that macrophages originating from monocytes can use either amoeboid or mesenchymal modes of migration depending on the structure of the surrounding matrix [168, 169]. In a matrix with heterogenic structure they could use both motility modes [170]. Leucocytes of other types migrated by the amoeboid mode [122, 168], and the ability to transition was not shown for them. Neutrophils used amoeboid motility during migration into collagen I gel [122]. Mouse neutrophils formed structures that looked like podosomes (the markers of mesenchymal motility), but neither existence of vinculin, typical for podosomes no protease activity were checked, so it could not be considered as evidence of mesenchymal migration [171]. It should be emphasized that directed migration of leucocytes to a site of inflammation occurs as a result of their activation, and thus ability for mutual MAT and AMT was shown for activated leucocytes. In our experiments studying the ability of transition of normal leucocytes and cells of chronic myeloid leukemia K562 for which amoeboid motility is typical, it was found that treatment of K562 cells with ROCK inhibitor Y27632 leads to development by the cells of adhesion structures with ECM and formation of lamellipodia (the signs of AMT), while the same treatment of normal leucocytes did not lead to such alterations (A. S. Chikina, unpublished).
PLASTICITY OF MIGRATION MECHANISMS OF STEM CELLS
Mutual EMT and the reverse MET are closely connected with cell migration during embryonic development. For gastrulation, pluripotent embryonic stem cells, which have epithelial characteristics of the inner embryo layer, go through EMT and develop to primary mesoderm [172]. Thus EMT is an initial differentiation step that leads to formation of three germ layers from pluripotent cells. Very often the transitions are associated with cell stemness. Expression of transcriptional factors SNAIL or TWIST, which is typical for EMT, induces the appearance of a special pool of mesenchymal cells carrying the stem markers CD44hiCD24low in a culture of breast epithelial cells [3].
In recent studies it was shown that synthesis of VEGF-C, which regulates angiogenesis in tumors, could regulate EMT and increase the number of tumor stem cells. Its suppression leads simultaneously both to reversion of EMT and to significant decrease in number of tumor cells in the population that could form colonies on semisolid medium [173]. It is important that cells as a result of EMT acquired typical features of tumor stem cells both on receptor levels and on the level of morphology and behavior [3, 4, 174]. These features are similar to those of normal stem cells [175]. For example, such common feature of normal and tumor stem cells is the ability to form podosomes in the process of mesenchymal migration. It was shown that trophoblasts that invade myometrium during placental development in early pregnancy form podosome-like structures [94]. It was also shown that stem cells often form podosomes during mesenchymal migration [176]. The ability of mutual MAT and AMT was shown for embryonic cells in course of development of Danio rerio (zebra fish, see above).
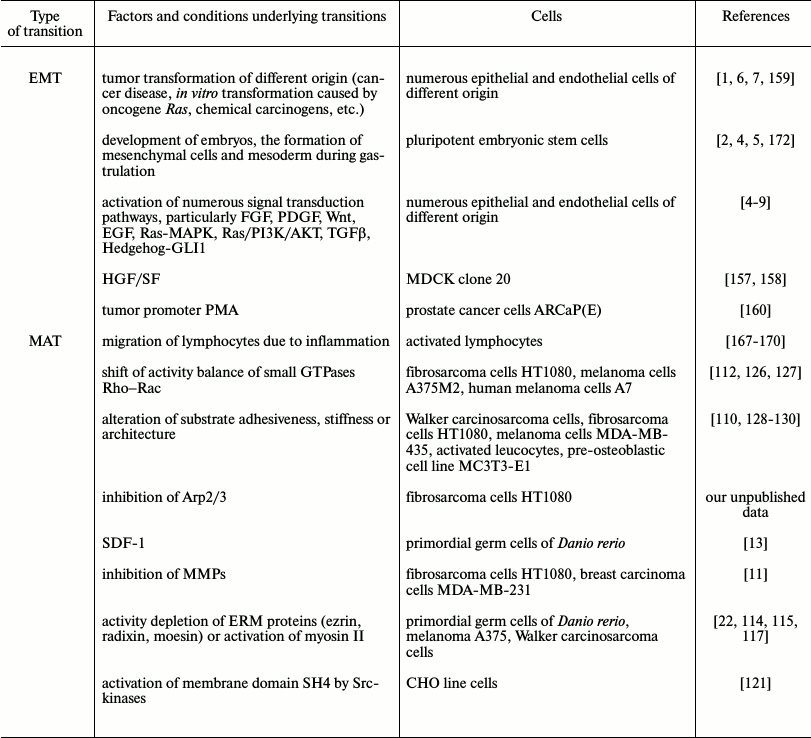
We summarized in the table the discussed data about the abilities for EMT and MAT transition regulating mechanisms of cell migration. One can see that plasticity of motility modes exists both in embryonic development an in tumor disease and, correspondingly, in experiments with embryonic or tumor cells. For normal differentiated cells plasticity, if any exists, is very limited.
External and inner factors causing EMT and MAT
The fact that tumor cells during cancer progression lose the features of differentiation and often begin to produce embryonal proteins, for example, α-fetoprotein, is long known and used in early diagnostics of tumor development [177]. Nevertheless, such abilities of cancer cells as invasion, disturbance of limit of cell divisions, increased production of MMPs for matrix degradation, and also acquisition of multi-drug resistance (MDR) due to active efflux of drugs from cells is a result of activity of cell proteins belonging to the superfamily of ABC-transporters [178], which was always believed characteristics of tumor cells that they acquire in the process of selection due to unlimited divisions and loss of control as a result, for example, of disturbance of the gene encoding p53 [179]. Recently it was shown that many of these features are typical for stem cells. Particularly the existence of active ABC-transporters is characteristic of different stem cells [180, 181], for hemopoietic stem cells, and for muscle satellite cells it was also shown that they could exclude the lipophilic dye Hoechst 33342 or rhodamine 123 [182]. Another feature of tumor cells that has been revealed in recent years is decreased stiffness of tumor cells. Stiffness is mainly determined by actin cytoskeleton, and decrease of stiffness is associated with increase in deformability, which probably could promote cell invasion to surrounding tissue and ECM and contribute to increase in dissemination [183, 184]. However, in other works it was shown that reduced stiffness and increased ability to deform are also features of stem cells [185, 186]. Analysis of the most recent works devoted to investigation of plasticity of migration mechanisms of tumor cells made in this review give us ideas that adaptations of cell migration widely used by tumor cells during invasion and metastasis and their plasticity are indeed using stem cell programs, which means a return to “well forgotten” old features.
The author thanks M. E. Lomakina and A. S. Chikina for kind permission to use their unpublished photographs and for helpful discussions.
This work was partially supported by the Russian Foundation for Basic Research grants Nos. 14-04-01228a and 14-04-91056.
REFERENCES
1.Hanahan, D., and Weinberg, R. A. (2011) Hallmarks
of cancer: the next generation, Cell, 144, 646-674.
2.Vasiliev, J. M. (2008) Reorganization of molecular
morphology of epitheliocytes and connective-tissue cells in
morphogenesis and carcinogenesis, Biochemistry (Moscow),
73, 528-531.
3.Mani, S. A., Guo, W., Liao, M. J., Eaton, E. N.,
Ayyanan, A., Zhou, A. Y., Brooks, M., Reinhard, F., Zhang, C. C.,
Shipitsin, M., Campbell, L. L., Polyak, K., Brisken, C., Yang, J., and
Weinberg, R. A. (2008) The epithelial-mesenchymal transition generates
cells with properties of stem cells, Cell, 133,
704-715.
4.Polyak, K., and Weinberg, R. A. (2009) Transitions
between epithelial and mesenchymal states: acquisition of malignant and
stem cell traits, Nature Rev. Cancer, 9, 265-273.
5.Lim, J., and Thiery, J. P. (2012)
Epithelial-mesenchymal transitions: insights from development,
Development, 139, 3471-3486.
6.Chaffer, C. L., Thompson, E. W., and Williams, E.
D. (2007) Mesenchymal to epithelial transition in development and
disease, Cells Tissues Organs, 185, 7-19.
7.Kopfstein, L., and Christofori, G. (2006)
Metastasis: cell-autonomous mechanisms versus contributions by the
tumor microenvironment, Cell Mol. Life Sci., 63,
449-468.
8.Lamouille, S., Xu, J., and Derynck, R. (2014)
Molecular mechanisms of epithelial-mesenchymal transition, Nature
Rev. Mol. Cell Biol., 15, 178-196.
9.Nelson, W. J. (2009) Remodeling epithelial cell
organization: transitions between front-rear and apical-basal polarity,
Cold Spring Harb. Perspect. Biol., 1, a000513.
10.Friedl, P., and Wolf, K. (2003) Proteolytic and
non-proteolytic migration of tumour cells and leucocytes, Biochem.
Soc. Symp., 70, 277-285.
11.Wolf, K., Mazo, I., Leung, H., Engelke, K., von
Andrian, U. H., Deryugina, E. I., Strongin, A. Y., Brocker, E. B., and
Friedl, P. (2003) Compensation mechanism in tumor cell migration:
mesenchymal-amoeboid transition after blocking of pericellular
proteolysis, J. Cell Biol., 20, 267-277.
12.Friedl, P., Hegerfeldt, Y., and Tusch, M. (2004)
Collective cell migration in morphogenesis and cancer, Int. J. Dev.
Biol., 48, 441-449.
13.Charras, G., and Paluch, E. (2008) Blebs lead the
way: how to migrate without lamellipodia, Nature Rev. Mol. Cell
Biol., 9, 730-736.
14.Fackler, O. T., and Grosse, R. (2008) Cell
motility through plasma membrane blebbing, J. Cell Biol.,
181, 879-884.
15.Hegerfeldt, Y., Tusch, M., Brocker, E. B., and
Friedl, P. (2002) Collective cell movement in primary melanoma
explants: plasticity of cell–cell interaction, beta-1-integrin
function, and migration strategies, Cancer Res., 62,
2125-2130.
16.Wolf, K., and Friedl, P. (2006) Molecular
mechanisms of cancer cell invasion and plasticity, Br. J.
Dermatol., 154, 11-15.
17.Pankova, K., Rosel, D., Novotny, M., and Brabek,
J. (2010) The molecular mechanisms of transition between mesenchymal
and amoeboid invasiveness in tumor cells, Cell Mol. Life Sci.,
67, 63-71.
18.Friedl, P., Zanker, K. S., and Brocker, E. B.
(1998) Cell migration strategies in 3D extracellular matrix:
differences in morphology, cell matrix interactions, and integrin
function, Microsc. Res. Tech., 43, 369-378.
19.Sahai, E., and Marshall, C. J. (2003) Differing
modes of tumour cell invasion have distinct requirements for Rho/ROCK
signaling and extracellular proteolysis, Nature Cell Biol.,
5, 711-719.
20.Friedl, P., Noble, P. B., Shields, E. D., and
Zanker, K. S. (1994) Locomotor phenotypes of unstimulated CD45RAhigh
and CD45ROhigh CD4+ and CD8+ lymphocytes in three-dimensional collagen
lattices, Immunology, 82, 617-624.
21.Niggemann, B., Maaser, K., Lu, H., Kroczek, R.,
Zanker, K. S., and Friedl, P. (1997) Locomotor phenotypes of human
tumor cell lines and T lymphocytes in a three-dimensional collagen
lattice, Cancer Lett., 118, 173-180.
22.Diz-Munoz, A., Krieg, M., Bergert, M.,
Ibarlucea-Benitez, I., Muller, D. J., Paluch, E., and Heisenberg, C. P.
(2010) Control of directed cell migration in vivo by
membrane-to-cortex attachment, PLoS Biol., 8,
e1000544.
23.Srinivasan, S., Wang, F., Glavas, S., Ott, A.,
Hofmann, F., Aktories, K., Kalman, D., and Bourne, H. R. (2003) Rac and
Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during
neutrophil chemotaxis, J. Cell Biol., 160,
375-385.
24.Sasaki, A. T., Chun, C., Takeda, K., and Firtel,
R. A. (2004) Localized Ras signaling at the leading edge regulates
PI3K, cell polarity, and directional cell movement, J. Cell
Biol., 167, 505-518.
25.Van Haastert, P. J., and Devreotes, P. N. (2004)
Chemotaxis: signalling the way forward, Nature Rev. Mol. Cell
Biol., 5, 626-634.
26.Charest, P. G., and Firtel, R. A. (2007) Big role
for small GTPases in control of directed cell movement, Biochem.
J., 401, 377-390.
27.Vorotnikov, A. V. (2011) Chemotaxis: movement,
direction, control, Biochemistry (Biochemistry), 76,
1528-1555.
28.Kraynov, V. S., Chamberlain, C., Bokoch, G. M.,
Schwartz, M. A., Slabaugh, S., and Hahn, K. M. (2000) Localized Rac
activation dynamics visualized in living cells, Science,
290, 333-337.
29.Itoh, R. E., Kurokawa, K., Ohba, Y., Yoshizaki,
H., Mochizuki, N., and Matsuda, M. (2002) Activation of rac and cdc42
video imaged by fluorescent resonance energy transfer-based
single-molecule probes in the membrane of living cells, Mol. Cell
Biol., 22, 6582-6591.
30.Schlunck, G., Damke, H., Kiosses, W. B., Rusk,
N., Symons, M. H., Waterman-Storer, C. M., Schmid, S. L., and Schwartz,
M. A. (2004) Modulation of Rac localization and function by dynamin,
Mol. Biol. Cell, 15, 256-267.
31.Haugh, J. M., Codazzi, F., Teruel, M., and Meyer,
T. (2000) Spatial sensing in fibroblasts mediated by 3′
phosphoinositides, J. Cell Biol., 151,
1269-1280.
32.Cote, J.-F., Motoyama, A. B., Bush, J. A., and
Vuori, K. (2005) A novel and evolutionarily conserved
PtdIns(3,4,5)P3-binding domain is necessary for DOCK180 signalling,
Nature Cell Biol., 7, 797-807.
33.Pollard, T. D., Blanchoin L., and Mullins, R. D.
(2000) Molecular mechanisms controlling actin filament dynamics in
nonmuscle cells, Annu. Rev. Biophys. Biomol. Struct., 29,
545-576.
34.Pollard, T. D. (2007) Regulation of actin
filament assembly by Arp2/3 complex and formins, Annu. Rev. Biophys.
Biomol. Struct., 36, 451-477.
35.Ridley, A. J., Schwartz, M. A., Burridge, K.,
Firtel, R. A., Ginsberg, M. H., Borisy, G., Parsons, J. T., and
Horwitz, A. R. (2003) Cell migration: integrating signals from front to
back, Science, 302, 1704-1709.
36.Burridge, K., and Doughman, R. (2006) Front and
back by Rho and Rac, Nature Cell Biol., 8, 781-782.
37.Pertz, O. L., Hodgson, R. L., Klemke, R. L., and
Hahn, K. M. (2006) Spatiotemporal dynamics of RhoA activity in
migrating cells, Nature, 440, 1069-1072.
38.Xu, J., Wang, F., Van Keymeulen, A., Herzmark,
P., Straight, A., Kelly, K., Takuwa, Y., Sugimoto, N., Mitchison, T.,
and Bourne, H. R. (2003) Divergent signals and cytoskeletal assemblies
regulate self-organizing polarity in neutrophils, Cell,
114, 201-214.
39.Van Hennik, P. B., ten Klooster, J. P., Halstead,
J. R., Voermans, C., Anthony, E. C., Divecha, N., and Hordijk, P. L.
(2003) The C-terminal domain of Rac1 contains two motifs that
control targeting and signaling specificity, J. Biol. Chem.,
278, 39166-39175.
40.Ten Klooster, J. P., Jaffer, Z. M., Chernoff, J.,
and Hordijk, P. L. (2006) Targeting and activation of Rac1 are mediated
by the exchange factor β-Pix, J. Cell Biol.,
172, 759-769.
41.Bass, M. D., Roach, K. A., Morgan, M. R.,
Mostafavi-Pour, Z., Schoen, T., Muramatsu, T., Mayer, U., Ballestrem,
C., Spatz, J. P., and Humphries, M. J. (2007) Syndecan-4-dependent Rac1
regulation determines directional migration in response to the
extracellular matrix, J. Cell Biol., 177,
527-538.
42.Pankov, R., Endo, Y., Even-Ram, S., Araki, M.,
Clark, K., Cukierman, E., Matsumoto, K., and Yamada, K. M. (2005) A Rac
switch regulates random versus directionally persistent cell migration,
J. Cell Biol., 170, 793-802.
43.Wu, Yi. I., Frey, D., Lungu, O. I., Jaehrig, A.,
Schlichting, I., Kuhlman, B., and Hahn, K. M. (2009) A
genetically-encoded photoactivatable Rac controls the motility of
living cells, Nature, 461, 104-108.
44.Vasiliev, J. M., Gelfand, I. M., Domnina, L. V.,
Ivanova, O. Y., Komm, S. G., and Olshevskaja, L. V. (1970) Effect of
colcemid on the locomotory behaviour of fibroblasts, J. Embryol.
Exp. Morphol., 24, 625-640.
45.Watanabe, T., Noritake, J., and Kaibuchi, K.
(2005) Regulation of microtubules in cell migration, Trends Cell
Biol., 15, 76-83.
46.Kaverina, I., and Straube, A. (2011) Regulation
of cell migration by dynamic microtubules, Semin. Cell Dev.
Biol., 22, 968-974.
47.Albiges-Rizo, C., Destaing, O., Fourcade, B.,
Planus, E., and Block, M. R. (2009) Actin machinery and
mechanosensitivity in invadopodia, podosomes and focal adhesions, J.
Cell Sci., 122, 3037-3049.
48.Mejillano, M. R., Kojima, S., Applewhite, D. A.,
Gertler, F. B., Svitkina, T. M., and Borisy, G. G. (2004) Lamellipodial
versus filopodial mode of the actin nanomachinery: pivotal role of the
filament barbed end, Cell Press, 118, 363-373.
49.Svitkina, T. M., Bulanova, T. A., Chaga, O. Y.,
Vignjevic, D. M., Kojima, Sh., Vasiliev, J. M., and Borisy, G. G.
(2003) Mechanisms of filopodia initiation by reorganization of a
dendritic network, J. Cell Biol., 160, 409.
50.Yang, C., Czech, L., Gerboth, S., Kojima, S.,
Scita, G., and Svitkina, T. (2007) Novel roles of formin mDia2 in
lamellipodia and filopodia formation in motile cells, PLoS
Biol., 5, 317.
51.Small, J. V., and Sechi, A. (1988) Whole-mount
electron microscopy of the cytoskeleton: negative staining methods, in
Cell Biology: a Laboratory Handbook (Celis, J. E., ed.) Vol. 3,
Academic Press, pp. 285-292.
52.Lewis, A. K., and Bridgman, P. C. (1992) Nerve
growth cone lamellipodia contain two populations of actin filaments
that differ in organization and polarity, J. Cell Biol.,
119, 1219-1243.
53.Small, J. V., Stradal, T., Vignal, E., and
Rottner, K. (2002) The lamellipodium: where motility begins, Trends
Cell Biol., 12, 112-120.
54.Mitchison, T. J., and Cramer, L. P. (1996)
Actin-based cell motility and cell locomotion, Cell, 84,
371-379.
55.Vignjevic, D., Kojima, S., Aratyn, Y., Danciu,
O., Svitkina, T., and Borisy, G. G. (2006) Role of fascin in filopodial
protrusion, J. Cell Biol., 174, 863-875.
56.Gupton, S. L., and Gertler, F. B. (2007)
Filopodia: the fingers that do the walking, Sci. STKE,
400.
57.Mattila, P. K., and Lappalainen, P. (2008)
Filopodia: molecular architecture and cellular functions, Nature
Rev. Mol. Cell Biol., 9, 446-454.
58.Yang, C., and Svitkina, T. (2011) Filopodia
initiation: focus on the Arp2/3 complex and formins, Cell Adh.
Migr., 5, 402-408.
59.Ridley, A. J. (2001) Rho GTPases and cell
migration, J. Cell Sci., 114, 2713-2722.
60.Bentley, D., and Toroian-Raymond, A. (1986)
Disoriented pathfinding by pioneer neuron growth cones deprived of
filopodia by cytochalasin treatment, Nature, 323,
712-715.
61.Chien, C. B., Rosenthal, D. E., Harris, W. A.,
and Holt, C. E. (1993) Navigational errors made by growth cones without
filopodia in the embryonic Xenopus brain, Neuron,
11, 237-251.
62.Davenport, R. W., Dou, P., Rehder, V., and Kater,
S. B. (1993) A sensory role for neuronal growth cone filopodia,
Nature, 361, 721-724.
63.Zheng, J. Q., Wan, J. J., and Poo, M. M. (1996)
Essential role of filopodia in chemotropic turning of nerve growth cone
induced by a glutamate gradient, J. Neurosci., 16,
1140-1149.
64.Grabham, P. W., Foley, M., Umeojiako, A., and
Goldberg, D. J. (2000) Nerve growth factor stimulates coupling of beta1
integrin to distinct transport mechanisms in the filopodia of growth
cones, J. Cell Sci., 113, 3003-3012.
65.Lanier, L. M., Gates, M. A., Witke, W., Menzies,
A. S., Wehman, A. M., Macklis, J. D., Kwiatkowski, D., Soriano, P., and
Gertler, F. B. (1999) Mena is required for neurulation and commissure
formation, Neuron, 22, 313-325.
66.Bear, J. E., Svitkina, T. M., Krause, M.,
Schafer, D. A., Loureiro, J. J., Strasser, G. A., Maly, I. V., Chaga,
O. Y., Cooper, J. A., and Borisy, G. G. (2002) Antagonism between
Ena/VASP proteins and actin filament capping regulates fibroblast
motility, Cell, 109, 509-521.
67.Tokuo, H., and Ikebe, M. (2004) Myosin X
transports Mena/VASP to the tip of filopodia, Biochem. Biophys. Res.
Commun., 319, 214-220.
68.Berg, J. S., and Cheney, R. E. (2002) Myosin-X is
an unconventional myosin that undergoes intrafilopodial motility,
Nature Cell Biol., 4, 246-250.
69.Mattila, P. K., Pykalainen, A., Saarikangas, J.,
Paavilainen, V. O., Vihinen, H., Jokitalo, E., and Lappalainen, P.
(2007) Missing-in-metastasis and IRSp53 deform PI(4,5)P2-rich membranes
by an inverse BAR domain-like mechanism, J. Cell Biol.,
176, 953-964.
70.Yang, C., Hoelzle, M., Disanza, A., Scita, G.,
and Svitkina, T. (2009) Coordination of membrane and actin cytoskeleton
dynamics during filopodia protrusion, PLoS One, 4,
e5678.
71.Pellegrin, S., and Mellor, H. (2005) The Rho
family GTPase Rif induces filopodia through mDia2, Curr.
Biol., 15, 129-133.
72.Mellor, H. (2010) The role of formins in
filopodia formation, Biochim. Biophys. Acta, 1803,
191-200.
73.Small, J. V., Isenberg, G., and Celis, J. E.
(1978) Polarity of actin at the leading edge of cultured cells,
Nature, 272, 638-639.
74.Small, J. V., Rottner, K., Kaverina, I., and
Anderson, K. I. (1998) Assembling an actin cytoskeleton for cell
attachment and movement, Biochim. Biophys. Acta, 1404,
271-481.
75.Chan, K. T., Cortesio, C. L., and Huttenlocher,
A. (2009) FAK alters invadopodia and focal adhesion composition and
dynamics to regulate breast cancer invasion, J. Cell Biol.,
185, 357-370.
76.Kovar, D. R., and Pollard, T. D. (2004)
Insertional assembly of actin filament barbed ends in association with
formins produces piconewton forces, Proc. Natl. Acad. Sci. USA,
101, 14725-14730.
77.Mullins, R. D., Heuser, J. A., and Pollard, T. D.
(1998) The interaction of Arp2/3 complex with actin: nucleation, high
affinity pointed end capping, and formation of branching networks of
filaments, Proc. Natl. Acad. Sci. USA, 95, 6181-6186.
78.Pollard, T. D., and Borisy, G. G. (2003) Cellular
motility driven by assembly and disassembly of actin filaments,
Cell, 112, 453-465.
79.Yamazaki, D., Kurisu, S., and Takenawa, T. (2005)
Regulation of cancer cell motility through actin reorganization,
Cancer Sci., 96, 379-386.
80.Stradal, T. E., and Scita, G. (2006) Protein
complexes regulating Arp2/3-mediated actin assembly, Curr. Opin.
Cell Biol., 18, 4-10.
81.Geiger, B., Bershadsky, A., Pankov, R., and
Yamada, K. M. (2001) Transmembrane extracellular
matrix–cytoskeleton crosstalk, Nature Rev. Mol. Cell
Biol., 2, 793-805.
82.Kaverina, I., Krylyshkina, O., and Small, J. V.
(2002) Regulation of substrate adhesion dynamics during cell motility,
Int. J. Biochem. Cell Biol., 34, 746-761.
83.Webb, D. J., Brown, C. M., and Horwitz, A. F.
(2003) Illuminating adhesion complexes in migrating cells: moving
toward a bright future, Curr. Opin. Cell Biol., 15,
614-620.
84.Sastry, S. K., and Burridge, K. (2000) Focal
adhesions: a nexus for intracellular signaling and cytoskeletal
dynamics, Exp. Cell. Res., 261, 25-36.
85.Zaidel-Bar, R., Itzkovitz, S., Ma’ayan, A.,
Iyengar, R., and Geiger, B. (2007) Functional atlas of the integrin
adhesome, Nature Cell Biol., 9, 858-867.
86.Takino, T., Watanabe, Y., Matsui, M., Miyamori,
H., Kodo, T., Seiki, M., and Sato, H. (2006) Membrane-type 1 matrix
metalloproteinase modulates focal adhesion stability and cell
migration, Exp. Cell Res., 312, 1381-1389.
87.Alexandrova, A. Y., Arnold, K., Schaub, S.,
Vasiliev, J. M., Meister, J. J., Bershadsky, A. D., and Verkhovsky, A.
B. (2008) Comparative dynamics of retrograde actin flow and focal
adhesions: formation of nascent adhesions triggers transition from fast
to slow flow, PLoS One, 3, e3234.
88.Choi, C. K., Vicente-Manzanares, M., Zareno, J.,
Whitmore, L. A., Mogilner, A., and Horwitz, A. R. (2008) Actin and
alpha-actinin orchestrate the assembly and maturation of nascent
adhesions in a myosin II motor-independent manner, Nature Cell
Biol., 10, 1039-1050.
89.Seals, D. F., Azucena, E. F., Jr., Pass, I.,
Tesfay, L., Gordon, R., Woodrow, M., Resau, J. H., and Courtneidge, S.
A. (2005) The adaptor protein Tks5/Fish is required for podosome
formation and function, and for the protease-driven invasion of cancer
cells, Cancer Cell, 7, 155-165.
90.Carman, C. V., Sage, P. T., Sciuto, T. E., de la
Fuente, M. A., Geha, R. S., Ochs, H. D., Dvorak, H. F., Dvorak, A. M.,
and Springer, T. A. (2007) Transcellular diapedesis is initiated by
invasive podosomes, Immunity, 26, 784-797.
91.Weaver, A. M. (2008) Invadopodia, Curr.
Biol., 18, 362-364.
92.Buccione, R., Caldieri, G., and Ayala, I. (2009)
Invadopodia: specialized tumor cell structures for the focal
degradation of the extracellular matrix, Cancer Metastasis Rev.,
28, 137-149.
93.Linder, S. (2007) The matrix corroded: podosomes
and invadopodia in extracellular matrix degradation, Trends Cell
Biol., 17, 107-117.
94.Patel, A., and Dash, P. R. (2012) Formation of
atypical podosomes in extravillous trophoblasts regulates extracellular
matrix degradation, Eur. J. Cell Biol., 91, 171-179.
95.Destaing, O., Saltel, F., Geminard, J. C.,
Jurdic, P., and Bard, F. (2003) Podosomes display actin turnover and
dynamic self-organization in osteoclasts expressing actin–green
fluorescent protein, Mol. Biol. Cell, 14, 407-416.
96.Nakahara, H., Howard, L., Thompson, E. W., Sato,
H., Seiki, M., Yeh, Y., and Chen, W. T. (1997)
Transmembrane/cytoplasmic domain-mediated membrane type 1-matrix
metalloprotease docking to invadopodia is required for cell invasion,
Proc. Natl. Acad. Sci. USA, 94, 7959-7964.
97.Redondo-Munoz, J., Escobar-Diaz, E., Samaniego,
R., Terol, M. J., Garcia-Marco, J. A., and Garcia-Pardo, A. (2006)
MMP-9 in B-cell chronic lymphocytic leukemia is upregulated by
α4β1 integrin or CXCR4 engagement via
distinct signaling pathways, localizes to podosomes, and is involved in
cell invasion and migration, Blood, 108, 3143-3151.
98.Sato, T., del Carmen Ovejero, M., Hou, P.,
Heegaard, A. M., Kumegawa, M., Foged, N. T., and Delaisse, J. M. (1997)
Identification of the membrane-type matrix metalloproteinase MT1-MMP in
osteoclasts, J. Cell Sci., 110, 589-596.
99.Coleman, M. L., Sahai, E. A., Yeo, M., Bosch, M.,
Dewar, A., and Olson, M. F. (2001) Membrane blebbing during apoptosis
results from caspase-mediated activation of ROCK I, Nature Cell
Biol., 3, 339-345.
100.Sebbagh, M., Renvoize, C., Hamelin, J., Riche,
N., Bertoglio, J., and Breard, J. (2001) Caspase-3-mediated cleavage of
ROCK I induces MLC phosphorylation and apoptotic membrane blebbing,
Nature Cell Biol., 3, 346-352.
101.Yoshida, K., and Soldati, T. (2006) Dissection
of amoeboid movement into two mechanically distinct modes, J. Cell
Sci., 119, 3833-3844.
102.Maugis, B., Brugues, J., Nassoy, P., Guillen,
N., Sens, P., and Amblard, F. (2010) Dynamic instability of the
intracellular pressure drives bleb-based motility, J. Cell Sci.,
123, 3884-3892.
103.Weiser, D. C., Row, R. H., and Kimelman, D.
(2009) Rho-regulated myosin phosphatase establishes the level of
protrusive activity required for cell movements during zebrafish
gastrulation, Development, 136, 2375-2384.
104.Cunningham, C. C. (1995) Actin polymerization
and intracellular solvent flow in cell surface blebbing, J. Cell
Biol., 129, 1589-1599.
105.Hagmann, J., Burger, M. M., and Dagan, D.
(1999) Regulation of plasma membrane blebbing by the cytoskeleton,
J. Cell. Biochem., 73, 488-499.
106.Charras, G. T., Yarrow, J. C., Horton, M. A.,
Mahadevan, L., and Mitchison, T. J. (2005) Non-equilibration of
hydrostatic pressure in blebbing cells, Nature,
435, 365-369.
107.Charras, G. T., Hu, C. K., Coughlin, M., and
Mitchison, T. J. (2006) Reassembly of contractile actin cortex in cell
blebs, J. Cell Biol., 175, 477-490.
108.Charras, G. T., Coughlin, M., Mitchison, T. J.,
and Mahadevan, L. (2008) Life and times of a cellular bleb, Biophys.
J., 94, 1836-1853.
109.Tinevez, J. Y., Schulze, U., Salbreux, G.,
Roensch, J., Joanny, J. F., and Paluch, E. (2009) Role of cortical
tension in bleb growth, Proc. Natl. Acad. Sci. USA, 106,
18581-18586.
110.Kitzing, T. M., Sahadevan, A. S., Brandt, D.
T., Knieling, H., Hannemann, S., Fackler, O. T., Grosshans, J., and
Grosse, R. (2007) Positive feedback between Dia1, LARG and RhoA
regulates cell morphology and invasion, Genes Dev., 21,
1478-1483.
111.Kitzing, T. M., Wang, Y., Pertz, O., Copeland,
J. W., and Grosse, R. (2010) Formin-like 2 drives amoeboid invasive
cell motility downstream of RhoC, Oncogene, 29,
2441-2448.
112.Sanz-Moreno, V., Gadea, G., Ahn, J., Paterson,
H., Marra, P., Pinner, S., Sahai, E., and Marshall, J. (2008) Rac
activation and inactivation control plasticity of tumor cell movement,
Cell, 135, 510-523.
113.Paluch, E. K., and Raz, E. (2013) The role and
regulation of blebs in cell migration, Curr. Opin. Cell Biol.,
25, 582-590.
114.Goudarzi, M., Banisch, T. U., Mobin, M. B.,
Maghelli, N., Tarbashevich, K., Strate, I., van den Berg, J., Blaser,
H., Bandemer, S., Paluch, E., Bakkers, J., Tolic-Norrelykke, I. M., and
Raz, E. (2012) Identification and regulation of a molecular module for
bleb-based cell motility, Dev. Cell, 23, 210-218.
115.Lorentzen, A., Bamber, J., Sadok, A.,
Elson-Schwab, I., and Marshall, C. J. (2011) An ezrin-rich, rigid
uropod-like structure directs movement of amoeboid blebbing cells,
J. Cell Sci., 124, 1256-1267.
116.Yanase, Y., Hide, I., Mihara, S., Shirai, Y.,
Saito, N., Nakata, Y., Hide, M., and Sakai, N. (2011) A critical role
of conventional protein kinase C in morphological changes of rodent
mast cells, Immunol. Cell Biol., 89, 149-159.
117.Martinelli, S., Chen, E. J., Clarke, F., Lyck,
R., Affentranger, S., Burkhardt, J. K., and Niggli, V. (2013)
Ezrin/radixin/moesin proteins and flotillins cooperate to promote
uropod formation in T cells, Front. Immunol., 4, 84.
118.Rossy, J., Gutjahr, M. C., Blaser, N.,
Schlicht, D., and Niggli, V. (2007) Ezrin/moesin in motile Walker 256
carcinosarcoma cells: signal-dependent relocalization and role in
migration, Exp. Cell Res., 313, 1106-1120.
119.Niggli, V., and Rossy, J. (2008)
Ezrin/radixin/moesin: versatile controllers of signaling molecules and
of the cortical cytoskeleton, Int. J. Biochem. Cell Biol.,
40, 344-349.
120.Gadea, G., de Toledo, M., Anguille, C., and
Roux, P. (2007) Loss of p53 promotes RhoA-ROCK-dependent cell migration
and invasion in 3D matrices, J. Cell Biol., 178,
23-30.
121.Tournaviti, S., Hannemann, S., Terjung, S.,
Kitzing, T. M., Stegmayer, C., Ritzerfeld, J., Walther, P., Grosse, R.,
Nicke, W., and Fackler, O. T. (2007) SH4-domain-induced plasma membrane
dynamization promotes bleb-associated cell motility, J. Cell
Sci., 120, 3820-3829.
122.Sabeh, F., Shimizu-Hirota, R., and Weiss, S. J.
(2009) Protease-dependent versus independent cancer cell invasion
programs: three-dimensional amoeboid movement revisited, J. Cell
Biol., 185, 11-19.
123.Ridley, A. J. (2011) Life at the leading edge,
Cell, 145, 1012-1022.
124.Wilkinson, S., Paterson, H. F., and Marshall,
C. J. (2005) Cdc42-MRCK and Rho-ROCK signalling cooperate in myosin
phosphorylation and cell invasion, Nature Cell Biol., 7,
255-261.
125.Wyckoff, J. B., Pinner, S. E., Gschmeissner,
S., Condeelis, J. S., and Sahai, E. (2006) ROCK- and myosin-dependent
matrix deformation enables protease-independent tumor-cell invasion
in vivo, Curr. Biol., 16, 1515-1523.
126.Ohta, Y., Hartwig, J. H., and Stossel, T. P.
(2006) FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to
control actin remodeling, Nature Cell Biol., 8,
803-814.
127.Yamazaki, D., Kurisu, S., and Takenawa, T.
(2009) Involvement of Rac and Rho signaling in cancer cell motility in
3D substrates, Oncogene, 28, 1570-1583.
128.Bergert, M., Chandradoss, S. D., Desai, R. A.,
and Paluch, E. (2012) Cell mechanics control rapid transitions between
blebs and lamellipodia during migration, Proc. Natl. Acad. Sci.
USA, 109, 14434-14439.
129.Cougoule, C., Goethem, E., Le Cabec, V., and
Lafouresse, F. (2012) Blood leukocytes and macrophages of various
phenotypes have distinct abilities to form podosomes and to migrate in
3D environments, Eur. J. Cell Biol., 91,
938-949.
130.Ehrbar, M., Sala, A., Lienemann, P., Ranga, A.,
Mosiewicz, K., Bittermann, A., Rizzi, S. C., Weber, F. E., and Lutolf,
M. P. (2011) Elucidating the role of matrix stiffness in 3D cell
migration and remodeling, Biophys. J., 100, 284-293.
131.Liu, Z., Yang, X., Chen, C., Liu, B., Ren, B.,
Wang, L., Zhao, K., Yu, S., and Ming, H. (2013) Expression of the
Arp2/3 complex in human gliomas and its role in the migration and
invasion of glioma cells, Oncol. Rep., 30, 2127-2136.
132.Zaman, M. H., Trapani, L. M., Sieminski, A. L.,
Mackellar, D., Gong, H., Kamm, R. D., Wells, A., Lauffenburger, D. A.,
and Matsudaira, P. (2006) Migration of tumor cells in 3D matrices is
governed by matrix stiffness along with cell-matrix adhesion and
proteolysis, Proc. Natl. Acad. Sci. USA, 103,
10889-10894.
133.Sales, A. D., Lobo, C. H., Carvalho, A. A.,
Moura, A. A., and Rodrigues, A. P. R. (2013) Structure, function, and
localization of aquaporins: their possible implications on gamete
cryopreservation, Genet. Mol. Res., 12, 6718-6732.
134.Papadopoulos, M. C., Saadoun, S., and Verkman,
A. S. (2008) Aquaporins and cell migration, Pflugers Arch.,
456, 693-700.
135.Loitto, V. M., Forslund, T., Sundqvist, T.,
Magnusson, K. E., and Gustafsson, M. (2002) Neutrophil leukocyte
motility requires directed water influx, J. Leukoc. Biol.,
71, 212-222.
136.Loitto, V. M., Karlsson, T., and Magnusson, K.
E. (2009) Water flux in cell motility: expanding the mechanisms of
membrane protrusion, Cell Motil. Cytoskeleton, 66,
237-247.
137.Saadoun, S., Papadopoulos, M. C., Hara-Chikuma,
M., and Verkman, A. S. (2005) Impairment of angiogenesis and cell
migration by targeted aquaporin-1 gene disruption, Nature,
434, 786-792.
138.Karlsson, T., Glogauer, M., Ellen, R. P.,
Loitto, V. M., Magnusson, K. E., and Magalhaes, M. A. (2011) Aquaporin
9 phosphorylation mediates membrane localization and neutrophil
polarization, J. Leukoc. Biol., 90, 963-973.
139.Karlsson, T., Lagerholm, B. C., Vikstrom, E.,
Loitto, V. M., and Magnusson, K. E. (2013) Water fluxes through
aquaporin-9 prime epithelial cells for rapid wound healing, Biochem.
Biophys. Res. Commun., 430, 993-998.
140.Chen, X. M., O’Hara, S. P., Huang, B. Q.,
Splinter, P. L., Nelson, J. B., and LaRusso, N. F. (2005) Localized
glucose and water influx facilitates Cryptosporidium parvum
cellular invasion by means of modulation of host cell membrane
protrusion, Proc. Natl. Acad. Sci. USA, 102,
6338-6343.
141.Loitto, V. M., Huang, C., Sigal, Y. J., and
Jacobson, K. (2007) Filopodia are induced by aquaporin-9 expression,
Exp. Cell Res., 313, 1295-1306.
142.Karlsson, T., Bolshakova, A., Magalhaes, M. A.
O., Loitto, V. M., and Magnusson, K.-E. (2013) Fluxes of water through
aquaporin 9 weaken membrane–cytoskeleton anchorage and promote
formation of membrane protrusions, PLoS One, 8,
e59901.
143.Huebert, R. C., Vasdev, M. M., Shergill, U.,
Das, A., Huang, B. Q., Charlton, M. R., LaRusso, N. F., and Shah, V. H.
(2010) Aquaporin-1 facilitates angiogenic invasion in the pathologic
neovasculature that accompanies cirrhosis, Hepatology,
52, 238-248.
144.Schwab, A., Fabian, A., Hanley, P. J., and
Stock, C. (2012) Role of ion channels and transporters in cell
migration, Physiol. Rev., 92, 1865-1913.
145.Saadoun, S., Papadopoulos, M. C., Davies, D.
C., Krishna, S., and Bell, B. A. (2002) Aquaporin-4 expression is
increased in oedematous human brain tumours, J. Neurol. Neurosurg.
Psychiatry, 72, 262-265.
146.Clucas, J., and Valderrama, F. (2014) ERM
proteins in cancer progression, J. Cell Sci., 127,
267-275.
147.Serrador, J. M., Alonso-Lebrero, J. L., del
Pozo, M. A., Furthmayr, H., Schwartz-Albiez, R., Calvo, J., Lozano, F.,
and Sanchez-Madrid, F. (1997) Moesin interacts with the cytoplasmic
region of intercellular adhesion molecule-3 and is redistributed to the
uropod of T lymphocytes during cell polarization, J. Cell
Biol., 138, 1409-1423.
148.Prag, S., Parsons, M., Keppler, M. D.,
Ameer-Beg, S. M., Barber, P., Hunt, J., Beavil, A. J., Calvert, R.,
Arpin, M., Vojnovic, B., and Ng, T. (2007) Activated ezrin promotes
cell migration through recruitment of the GEF Dbl to lipid rafts and
preferential downstream activation of Cdc42, Mol. Biol. Cell,
18, 2935-2948.
149.Estecha, A., Sanchez-Martin, L., Puig-Kroger,
A., Bartolome, R. A., Teixido, J., Samaniego, R., and Sanchez-Mateos,
P. (2009) Moesin orchestrates cortical polarity of melanoma tumour
cells to initiate 3D invasion, J. Cell Sci., 122,
3492-3501.
150.Khanna, C., Wan, X., Bose, S., Cassaday, R.,
Olomu, O., Mendoza, A., Yeung, C., Gorlick, R., Hewitt, S. M., and
Helman, L. J. (2004) The membrane–cytoskeleton linker ezrin is
necessary for osteosarcoma metastasis, Nature Med.,
10, 182-186.
151.Yu, Y., Khan, J., Khanna, C., Helman, L.,
Meltzer, P. S., and Merlino, G. (2004) Expression profiling identifies
the cytoskeletal organizer ezrin and the developmental homeoprotein
Six-1 as key metastatic regulators, Nature Med.,
10, 175-181.
152.Kobayashi, H., Sagara, J., Kurita, H.,
Morifuji, M., Ohishi, M., Kurashina, K., and Taniguchi, S. (2004)
Clinical significance of cellular distribution of moesin in patients
with oral squamous cell carcinoma, Clin. Cancer Res., 10,
572-580.
153.Condeelis, J., Singer, R. H., and Segall, J. E.
(2005) The great escape: when cancer cells hijack the genes for
chemotaxis and motility, Annu. Rev. Cell Dev. Biol.,
21, 695-718.
154.Charafe-Jauffret, E., Monville, F., Bertucci,
F., Esterni, B., Ginestier, C., Finetti, P., Cervera, N., Geneix, J.,
Hassanein, M., Rabayrol, L., Sobol, H., Taranger-Charpin, C., Xerri,
L., Viens, P., Birnbaum, D., and Jacquemier, J. (2007) Moesin
expression is a marker of basal breast carcinomas, Int. J.
Cancer, 121, 1779-1785.
155.Hiscox, S., and Jiang, W. G. (1999) Ezrin
regulates cell–cell and cell–matrix adhesion, a possible
role with E-cadherin/beta-catenin, J. Cell Sci.,
112, 3081-3090.
156.Fehon, R. G., McClatchey, A. I., and Bretscher,
A. (2010) Organizing the cell cortex: the role of ERM proteins, Rev.
Mol. Cell Biol., 11, 276-287.
157.Dugina, V. B., Alexandrova, A., Lane, K.,
Bulanova, E., and Vasiliev, J. M. (1995) The role of the microtubular
system in response to HGF/SF, J. Cell Sci., 108,
1659-1667.
158.Laser-Azogui, A., Diamant-Levi, T., Israeli,
S., Roytman, Y., and Tsarfaty, I. (2013) Met-induced membrane blebbing
leads to amoeboid cell motility and invasion, Oncogene,
33, 1-11.
159.Alexandrova, A. Y., Dugina, V. B., Ivanova, O.
J., Kaverina, I. N., and Vasiliev, J. M (1998) Scatter factor
induces segregation of multinuclear cells into several discrete motile
domains, Cell Motil. Cytoskeleton, 39, 147-158.
160.Zhitniak, I. Iu., and Glushankova, N. A. (2011)
Morphology, cell–cell interactions, and migratory activity of
IAR-2 epithelial cells transformed with the RAS oncogene: contribution
of cell adhesion protein E-cadherin, Ontogenez, 42,
453-464.
161.He, H., Davidson, A. J., Wu, D., Marshall, F.
F., Chung, L. W., Zhau, H. E., He, D., and Wang, R. (2010) Phorbol
ester phorbol-12-myristate-13-acetate induces epithelial to mesenchymal
transition in human prostate cancer ARCaPE cells, Prostate,
70, 1119-1126.
162.Niggemann, B., Drell, T. L., Joseph, J., Weidt,
C., Lang, K., Zaenker, K. S., and Entschladen, F. (2004) Tumor cell
locomotion: differential dynamics of spontaneous and induced migration
in a 3D collagen matrix, Exp. Cell Res., 298,
178-187.
163.Artym, V. V., Zhang, Y., Seillier-Moiseiwitsch,
F., Yamada, K. M., and Mueller, S. C. (2006) Dynamic interactions of
cortactin and membrane type 1 matrix metalloproteinase at invadopodia:
defining the stages of invadopodia formation and function, Cancer
Res., 66, 3034-3043.
164.Buccione, R., Orth, J. D., and McNiven, M. A.
(2004) Foot and mouth: podosomes, invadopodia and circular dorsal
ruffles, Nature Rev. Mol. Cell Biol., 5, 647-657.
165.Hauck, C. R., Hsia, D. A., Ilic, D., and
Schlaepfer, D. D. (2002) v-Src SH3-enhanced interaction with focal
adhesion kinase at beta 1 integrin-containing invadopodia promotes cell
invasion, J. Biol. Chem., 277, 12487-12490.
166.Singh, V. P., and McNiven, M. A. (2008)
Src-mediated cortactin phosphorylation regulates actin localization and
injurious blebbing in acinar cells, Mol. Biol. Cell, 19,
2339-2347.
167.Friedl, P., Borgmann, S., and Brocker, E. B.
(2001) Amoeboid leukocyte crawling through extracellular matrix:
lessons from the Dictyostelium paradigm of cell movement, J.
Leukoc. Biol., 70, 491-509.
168.Friedl, P., and Weigelin, B. (2008)
Interstitial leukocyte migration and immune function, Nature
Immunol., 9, 960-969.
169.Van Goethem, E., Poincloux, R., Gauffre, F.,
Maridonneau-Parini, I., and Le Cabec, V. (2010) Matrix architecture
dictates three-dimensional migration modes of human macrophages:
differential involvement of proteases and podosome-like structures,
J. Immunol., 184, 1049-1061.
170.Guiet, R., Van Goethem, E., Cougoule, C.,
Balor, S., Valette, A., Al Saati, T., Lowell, C. A., Le Cabec, V., and
Maridonneau-Parini, I. (2011) The process of macrophage migration
promotes matrix metalloproteinase-independent invasion by tumor cells,
J. Immunol., 187, 3806-3814.
171.Szczur, K., Xu, H., Atkinson, S., Zheng, Y.,
and Filippi, M. D. (2006) Rho GTPase CDC42 regulates directionality and
random movement via distinct MAPK pathways in neutrophils,
Blood, 108, 4205-4213.
172.Acloque, H., Adams, M. S., Fishwick, K.,
Bronner-Fraser, M., and Nieto, M. A. (2009) Epithelial-mesenchymal
transitions: the importance of changing cell state in development and
disease, J. Clin. Invest., 119, 1438-1449.
173.Khromova, N., Kopnin, P., Rybko, V., and
Kopnin, B. P. (2012) Downregulation of VEGF-C expression in lung and
colon cancer cells decelerates tumor growth and inhibits metastasis via
multiple mechanisms, Oncogene, 31, 1389-1397.
174.Singh, A., and Settleman, J. (2010) EMT, cancer
stem cells and drug resistance: an emerging axis of evil in the war on
cancer, Oncogene, 29, 4741-4751.
175.Chikina, A. S., and Alexandrova, A. Y. (2014)
The cellular mechanisms and regulation of metastasis formation, Mol.
Biol. (Moscow), 48, 165-180.
176.Smith, H., Whittall, C., Weksler, B., and
Middleton, J. (2012) Chemokines stimulate bidirectional migration of
human mesenchymal stem cells across bone marrow endothelial cells,
Stem Cells, 21, 476-486.
177.Abelev, G. I. (1989) Alpha-fetoprotein: 25
years of study, Tumour Biol., 10, 63-74.
178.Stavrovskaya, A. A., and Stromskaya, T. P.
(2008) Transport proteins of the ABC family and multidrug resistance of
tumor cells, Biochemistry (Moscow), 73, 592-604.
179.Chumakov, P. M. (2007) Versatile functions of
p53 protein in multicellular organisms, Biochemistry (Moscow),
72, 1399-1421.
180.Zhou, S., Schuetz, J. D., Bunting, K. D.,
Colapietro, A. M., Sampath, J., Morris, J. J., Lagutina, I., Grosveld,
G. C., Osawa, M., Nakauchi, H., and Sorrentino, B. P. (2001) The ABC
transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells
and is a molecular determinant of the side-population phenotype,
Nature Med., 7, 1028-1034.
181.Bunting, K. D. (2002) ABC transporters as
phenotypic markers and functional regulators of stem cells, Stem
Cells, 20, 11-20.
182.Paltsev, M. A. (ed.) (2009) Biology of Stem
Cells and Cellular Technologies [in Russian], Meditsina,
Moscow.
183.Zhang, W., Kai, K., Choi, D. S., Iwamoto, T.,
Nguyen, Y. H., Wong, H., Landis, M. D., Ueno, N. T., Chang, J., and
Qin, L. (2012) Microfluidics separation reveals the stem-cell-like
deformability of tumor-initiating cells, Proc. Natl. Acad. Sci.
USA, 109, 18707-18712.
184.Efremov, Y. M., Lomakina, M. E., Bagrov, D. V.,
Makhnovskiy, P. I., Alexandrova, A. Y., Kirpichnikov, M. P., and
Shaitan, K. V. (2014) Mechanical properties of fibroblasts depend on
level of cancer transformation, Biochim. Biophys. Acta,
1843, 1013-1019.
185.Pajerowski, J. D., Dahl, K. N., Zhong, F. L.,
Sammak, P. J., and Discher, D. E. (2007) Physical plasticity of the
nucleus in stem cell differentiation, Proc. Natl. Acad. Sci.
USA, 104, 15619-15624.
186.Titushkin, I., and Cho, M. (2007) Modulation of
cellular mechanics during osteogenic differentiation of human
mesenchymal stem cells, Biophys. J., 93, 3693-3702.