REVIEW: New Data on Programmed Aging – Slow Phenoptosis
M. V. Skulachev1,2 and V. P. Skulachev2,3*
1Biological Faculty, Lomonosov Moscow State University, 119991 Moscow, Russia2Institute of Mitoengineering, Lomonosov Moscow State University, 119991 Moscow, Russia
3Belozersky Institute of Physico-Chemical Biology and Department of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: skulach@belozersky.msu.ru; skulach@genebee.msu.ru
* To whom correspondence should be addressed.
Received June 11, 2014
This review summarizes the latest data on biochemistry and physiology of living organisms. These data suggest that aging, i.e. coordinated age-dependent weakening of many vital functions leading to gradual increase in the probability of dying, is not common to all organisms. Some species have been described whose probability of death does not depend on age or even decreases with age, this being accompanied by constant or increasing fertility. In the case of the naked mole rat (a non-aging mammal), a mechanism has been identified that protects this animal from cancer and the most common age-related diseases. The high molecular weight polysaccharide hyaluronan, a linear polymer composed of multiple repeated disaccharide of glucuronic acid and glucosamine, plays the key role in this mechanism. Hyaluronan is accumulated in the intercellular spaces in the organs and tissues of the naked mole rat. This polysaccharide provides early contact inhibition of cell division (anti-cancer effect). In addition, hyaluronan prevents the development of certain types of apoptosis, in particular, those induced by reactive oxygen species (ROS) (geroprotective effect preventing ROS-induced decrease in cellularity in the organs and tissues of aging organisms). Extraordinary longevity of the naked mole rat (over 30 years, which is long for a rodent the size of a mouse) is connected to its eusocial lifestyle, when only the “queen” and its few “husbands” breed, while the huge army of non-breeding “subordinates” provide the “royal family” with protection from predators, food, and construction and maintenance of an underground labyrinth size of a football field. This way of life removes the pressure of natural selection from the “family” and makes aging – the program that is counterproductive for the individual but increases “evolvability” of its offspring – unnecessary. The example of the naked mole rat demonstrates the optional character of the aging program for the organism. Many facts indicating that aging can be regulated by an organism provide another argument in favor of optionality of aging. Cases have been described when aging as a program useful for the evolution of offspring but counterproductive for the parental individual slows under conditions that threaten the very existence of the individual. These conditions include food restriction (the threat of death from starvation), heavy muscular work, decrease or increase in the environmental temperature, small amounts of poisons (including ROS; here we speak about the paradoxical geroprotective effect of the low doses of prooxidants that inhibit apoptosis). On the other hand, aging can be inhibited (and maybe even cancelled) artificially. This can be done by turning off the genes encoding the proteins participating in the aging program, such as FAT10, p66shc, and some others. In addition, the gene of the antioxidant enzyme catalase can be addressed into mitochondria, where it will split mitochondrial hydrogen peroxide, the level of which increases with age. However, today the simplest way to slow down the aging program is the use of mitochondria-targeted low molecular weight antioxidant compounds of plastoquinonyl decyltriphenylphosphonium-type (SkQ1), which prolong the life of animals, plants, and fungi and inhibit the development of many age-related diseases and symptoms.
KEY WORDS: phenoptosis, aging program, geroprotectors, antioxidants, mitochondria, evolutionDOI: 10.1134/S0006297914100010
Abbreviations: mROS mitochondrial reactive oxygen species; ROS, reactive oxygen species; SkQ, derivatives of plastoquinone and penetrating cations (Sk+); SkQ1, plastoquinonyl decyltriphenylphosphonium.
It seems striking that complex multicellular organisms, having accomplished an obviously marvelous feat of morphogenesis, should not be able to solve a much simpler task of maintaining what has been already achieved.
G. Williams
To provide some explanation to Williams’ paradox [1], advocates of the concept of aging as the result of accumulation of random errors are forced to ascribe it to some defect of biological evolution that allowed such a drawback. Unfortunately, this postulate cannot be verified. However, before accusing evolution in imperfection, let us remember the advice of the great Francis Crick: the biologist should be guided by a simple rule – evolution is always smarter than he is.
The concept of aging as a special biological program provides an alternative to the hypothesis of random errors. According to this concept, aging is a particular case of the phenomenon of programmed death of an organism, phenoptosis [2-7]. Aging is assumed to accelerate evolution since over the years the organism weakened by aging is subjected to increasing pressure of natural selection. For example, a fox is hardly a factor of natural selection for young hares, which run much faster than the predator. As noted by Aesop, a hare will always run away from the fox because for the hare it is a matter of life and death, and for the fox – of a dinner. However, age-related sarcopenia reduces the hare’s running speed, so the fox gets a chance to win the race. As sarcopenia is one of the early signs of aging in mammals, developing well before senile infertility, foxes could accelerate the evolution of hares by eliminating the slowest and least clever individuals [8].
The biological literature contains many examples of phenoptosis enhancing the organism’s ability to evolve (their “evolvability”). Along with aging, they include different mechanisms providing, on one hand, increase in offspring diversity (which is beneficial for the search for new properties) and, on the other hand, the conservatism of inheriting of already acquired useful traits (for details, see [8]). These mechanisms, while being undoubtedly useful for evolution, are often counterproductive for the individual, as in the case of aging.
In this review, we will consider some recent studies indicating that aging is nothing but slow, multistage phenoptosis.
“NOAH’S ARK” OF PROFESSOR OWEN JONES
O. Jones et al. published an article in Nature on January 9, 2014 [9] where they compared the age-related curves of death probability and fertility for 46 different eukaryotic species: 11 species of mammals, 12 other vertebrates, 10 invertebrates, and 13 plants. As expected, the majority of the best-studied species, including humans, chimpanzees, baboons, lions, fruit flies, and the nematode Caenorhabditis elegans are characterized by age-related increase in mortality and decrease in fertility. However, the study has identified groups of species that are clearly not subject to this rule. For example, mortality and fertility are practically independent of age in hydra (Hydra magnipapillata), long-clawed hermit crab (Pagurus longicarpus), collared flycatcher (Ficedula albicollis), rhododendron (Rhododendron maximum), red-legged frog (Rana aurora), and red abalone (Haliotis rufescens). Fertility was shown to grow against increasing mortality in roe deer (Capreolus capreolus), Scotch pine (Pinus sylvestris), and the palm Geonoma orbignyana. Sometimes mortality may increase while fertility stays the same. The yellow baboon (Papio cynocephalus), Alpine swift (Apus melba), and body louse (Pediculus humanus) are examples of this phenomenon. Reliable cases have been described when mortality remained constant and fertility increased in the course of aging. These cases include yellow-bellied marmot (Marmota flaviventris), tundra vole (Microtus oeconomus), great tit (Parus major), mountain grass borderia (Borderia pyrenaica), highlands scrub hypericum (Hypericum cumulicola), and tubercled saltbush (Atriplex acanthocarpa). However, the most striking are the cases when mortality decreases and fertility increases with age (so-called “negative aging”). This phenomenon was found in desert tortoise (Gopherus agassizii), Mexican netleaf oak tree (Quercus rugosa), brown alga Oarweed (Laminaria digitata), forked viburnum (Viburnum furcatum), and red coral (Paramuricea clavata).
It is noteworthy that the variability of age-related dependences is not affected by the position of species on the evolutionary tree: it can be found in representatives of various classes of vertebrate and invertebrate animals as well as plants. Unfortunately, there is no naked mole rat in the “Jones list”. There are no data on the age-related dynamics of fertility for the naked mole rat, and the calibration of the ordinate in the single published chart of mortality as the function of age is rather questionable according to Jones et al. [9]. However, there is no doubt that this small rodent lives much longer than mice (over 32 years) and does not suffer from cancer, cardiovascular diseases, diabetes, and other pathologies lethal for most mammals. A clear technical advantage of the naked mole rat compared to ageless species from the “Jones list” is that it is a small mammal that easily endures life in the vivarium.
Jones et al. have obtained another important result, the variety of ageless species in terms of the level of complexity of their organisms and maximal lifespan. It seems especially important that there are examples of ageless organisms whose fertility increases with age, i.e. they live not only long, but also a literally fruitful life.
In general, the study by O. Jones et al. is a powerful new argument in favor of the idea on the optionality of aging for living organisms. It is perfectly consistent with the concept of aging as a facultative biological program used by species to accelerate their evolution under conditions requiring such acceleration.
THE NAKED MOLE RAT: SOLUTION OF THE SEARING MYSTERY BY V.
GORBUNOVA AND A. SELUANOV
The naked mole rat is a unique mammal (a rodent) the size of a mouse. Its maximal lifespan is at least eight times longer than that of mice. There are two important observations concerning these animals: 1) no cases of cancer have been described among them; 2) there are practically no senile diseases among the causes of their death. In 2013, V. Gorbunova, A. Seluanov, and their coworkers from the University of Rochester, USA, discovered the specific feature of the naked mole rat which could explain both of the these amazing properties and as a result the extraordinary longevity of this African animal [10].
They found that in case of the naked mole rat, hyaluronan, an extracellular unbranched polysaccharide consisting of dimers of glucuronic acid and N-acetylglucosamine, has several unique characteristics: a) its molecular weight is five times higher than that of mice, guinea pigs, or humans; b) it is formed faster by hyaluronan synthase-2 due to a change in the primary structure of this enzyme; c) it degrades more slowly due to reduced activity of the enzymes cleaving it; d) it binds stronger to the signaling hyaluronan receptors on the cell surface; e) the signal transmitted by high molecular weight hyaluronan bound to the receptor is the opposite from the signal from low molecular weight hyaluronans. It inhibits mitosis and prevents inflammation, while small hyaluronans stimulate both mitosis and inflammation. Large hyaluronans are the key component in the early contact inhibition of cell proliferation, the phenomenon discovered by the same group, which explains the nature of the naked mole rat’s resistance to cancer [11]. Strikingly, the same compounds were shown to be responsible for the prevention of also other age-related pathologies, and hence for the absence of aging in these animals, whose probability of death does not increase with age.
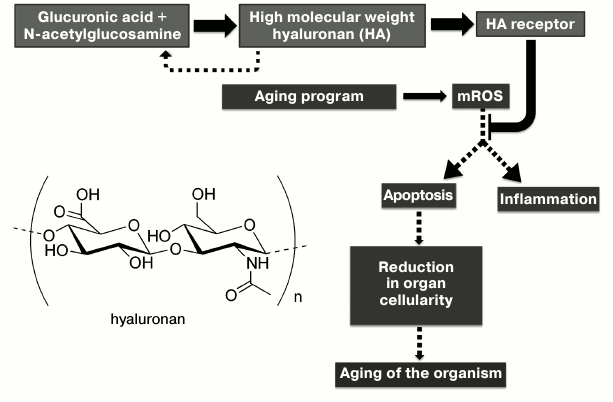
Simultaneously, another mystery of the naked mole rat was solved: its cells do not undergo apoptosis when hydrogen peroxide is added to cell culture. According to V. Gorbunova (personal communication), regular washing away of hyaluronan from naked mole rat cells is sufficient for H2O2 to turn into an inducer of their apoptosis. The antiapoptotic effect of hyaluronan can be explained by both its direct antioxidant (characteristic of carbohydrates) and the violation of the apoptotic cascade caused by ROS (reactive oxygen species) signal sent inside the cell by the receptor that binds hyaluronan (keep in mind the antiinflammatory effect of large hyaluronans)1. The second explanation provides the key to understanding another paradox of the naked mole rat: the levels of mitochondrial reactive oxygen species (mROS) and peroxidation products are higher in these animals than in mice; as a result, they do not follow the rule “formation of large amounts of mROS means short life” [14]. We can assume that the apoptotic cascade in the naked mole a rat is switched off somewhere after ROS and the products of cellular components oxidation by ROS. Inhibition of apoptosis by hyaluronan prevents the reduction of cellularity in organs and tissues, thus inhibiting the aging program (Fig. 1).
1 Our group has shown that hydrogen peroxide formed inside mitochondria is involved in the transmission of the apoptotic signal between cells [12, 13]. Excretion of H2O2 into the intercellular space leads to the “multiplication” of the apoptotic signal generated by the cell that has increased its ROS level. In this way, the signal is transmitted from cell to cell, causing their mass death. We assume that this effect can be prevented by large amounts of antioxidant (hyaluronan) in the intercellular space.
Fig. 1. Proposed mechanism of retardation of aging in naked mole rats: the role of high molecular weight hyaluronan (HA) and mROS. Thick arrows indicate the processes stimulated in naked mole rats, and dotted lines correspond to the inhibited processes.
At the end of October 2013, Gorbunova and Seluanov published another article [15], this time about the discovery of unusual properties of the structure and form of the naked mole rat ribosome. They found that the RNA molecule of the large (28S) subunit of these ribosomes is cleaved into two parts; a unique ribonucleotide sequence consisting of 118 nucleotides is linked to one of those parts in the cleavage site. The modified ribosome of the naked mole rat forms a protein with the same rate as a regular ribosome, but it makes far fewer errors when reading the informational RNA. Similar (but not identical) ribosomal structure was previously found in the South African rat-like rodent tuco-tuco (Ctenomys). The accuracy of the functioning of the ribosome in this animal was shown to be intermediate between that of the naked mole rat and the mouse (V. Gorbunova, personal communication). Tuco-tuco, similar to the naked mole rats, lives in large colonies (up to 200 animals), but each pair occupies a separate hole where they breed. As a result, their reproductive abilities are not monopolized by the queen as in the case of the naked mole rat. The lifespan of tuco-tuco is only about 3 years, which is more than 10 times shorter than that of the naked mole rat. (You can read about another distinctive property of the naked mole rat, the absence of one of the pain receptors, in an article by M. V. Skulachev, F. F. Severin, and V. P. Skulachev in this issue.)
OTHER LONGEVITY MECHANISMS
If aging is programmed, there should be different ways to cancel it because this program can be inhibited at different stages. It makes sense to compare the naked mole rats with bats and American white-footed mice, which also have very long lifespan.
Among the 10,000 species of bats, there are species that live up to 45 years with the aging program apparently completely switched off. In the case of the white-footed mouse (that lives for about 8 years), this program is probably only inhibited. In both bats [16, 17] and the white-footed mouse [18-21], mitochondria form ROS much more slowly than in other small mammals with shorter lifespans. Quantitatively, the lifespan of the white-footed mice is inversely related to the rate of mROS generation on reverse electron transfer in the respiratory chain. This dependence was first shown by Lambert et al. for 11 species of mammals and birds. The naked mole rat proved to be the only exception [14]. This paradox can be explained if we assume that the aging program in the naked mole rat is inhibited after the stage of mROS accumulation, namely, at the level of neutralization of extracellular H2O2 by hyaluronan and inhibition of ROS-dependent apoptosis. As for the bats and white-footed mice, this inhibition apparently takes place before mROS accumulation. In accordance with this hypothesis, it was shown that the antioxidant enzymes glutathione peroxidase, superoxide dismutase, and catalase are more active in the white-footed mice than in house mice [18, 20, 22].
In 1990, C. E. Finch suggested the existence of species with negligible aging [23, 24]. As a rule, these organisms have very long lifespan and are not subject to cancer, cardiovascular diseases, diabetes, and other senile ailments. They die of some causes independent of age. For example, the naked mole rats die because of fights with each other (these animals cannot be kept in isolation in an ordinary cage; when in captivity, they should live at least in small groups). This does not mean that senile pathologies are completely absent in naked mole rats. Some are occasionally observed, but they almost never lead to death [25]. It seems that these rodents, unlike the majority of other animals, when in a critical situation, do not undergo acute phenoptosis to rid the population of weakened individuals. The absence of both slow (aging) and at least one type of acute phenoptosis emphasizes the key feature of the naked mole rat – it has occupied an ecological niche where the species has no enemies (and therefore no pressure of natural selection). Only the queen and her few “husbands” breed, being protected and serviced by an army of their non-breeding offspring that cannot pass down to subsequent generations any acquired trait even if it would be useful.
The absence of enemies is also typical for many other non-aging animals protected from enemies by their impressive size or fighting qualities (large crabs and turtles, pikes, crocodiles, eagles, albatrosses2), inedibility (sea urchins, toads), special mechanisms of early danger warning (bats with their echolocation) or, finally, the high level of social organization (naked mole rats). These species already do not have any advantage from the enhancement of their further evolution and as a result they have lost phenoptotic programs providing such enhancement but counterproductive for individuals. It seems very important that in all the non-aging organisms examined for malignant diseases, cases of cancer were either detected very rarely, or were not detected at all (as, for example, in the naked mole rats) [25, 27]. This fact may indicate that cancer, as well as other senile diseases, is nothing but another phenoptotic program enhancing evolution (see below).
2 According to Lecomte et al. [26], aged albatrosses (lifespan over 50 years) nesting on the islands in the Indian Ocean can fly in search of prey into Antarctic waters, while young and middle-aged individuals never reach the Antarctic Circle (observations conducted by satellite and with radio sensors fixed on the birds). The authors were searching for any signs of aging in albatrosses, which have never been found in these animals growing through their entire life and suddenly dying for unknown reasons. Lecomte et al. came to conclusion that the age-related increase in flight distance is a sign of aging in albatrosses, which is of course contrary to the very definition of aging as the weakening of vital functions with age, and not their enhancement [8].
HOW TO RECONCILE SUPPORTERS OF CONCEPTS OF PROGRAMMED AGING AND
AGING AS ACCUMULATION OF RANDOM ERRORS?
The great physicist Leo Szilard believed the reduction of tissue and organ cellularity to be the main cause of aging [28]. According to Szilard, the problem of aging is not so much connected to the fact that each of our cells works worse, but that the number of these cells dramatically decreases with time. Senile sarcopenia, i.e. the reduction of the number of cells (myofibrils) in skeletal muscles, is a typical example of this phenomenon. The implementation of the aging program of skeletal muscles results in the organism demanding the performance of the same muscle functions despite decreasing myofibril number. Apparently, aging of the majority of our other tissues looks similar to this process (see the article by G. Libertini in this issue of Biochemistry (Moscow)). It is reminiscent of the policy of a cunning manufacturer who forces the factory to produce the same amount of goods while reducing the number of workers. To meet the challenge, the workers have only one possibility: to come up with something new and increase productivity. How is it possible to achieve this result with respect to the living cell?
Everything we have learned over the years about cells and their functioning undoubtedly convinces us of the extreme “bureaucratization” of its management. Long hierarchical chains of regulators have been found in cells. If an enzyme (e.g. muscle ATPase actomyosin) performs some useful work, a whole chain of other enzymes will monitor this process. This chain consists of the regulator N1 directly interacting with actomyosin; regulator 2, controlling the work of regulator N1; regulator 3 regulating regulator N2, and, finally, regulator 4, which controls the regulator 3 and at the same time is regulated by a hormone, the command device of the supracellular level. The amount of hormone in blood is, in turn, regulated by the chain of other regulators. This cumbersome system, when acting cohesively, increases the reliability of cell functioning, in particular by reducing the probability of accumulation of errors in DNA and protein structures. If such errors do occur, in DNA they are usually corrected by special systems of monitoring and reparation. As for damaged proteins, they are recognized and labeled by special enzymes monitoring the quality of these polymers. Special polypeptides, ubiquitins, are attached to such damaged proteins. Ubiquitinated proteins are recognized by the “molecular mincer”, the proteasome, a minute tube-like intracellular organelle that binds the proteins to be eliminated and splits them into constituent amino acids [29].
High reliability of the system of DNA and protein quality control is provided by the well-known redundancy of their work: ubiquitination removes not only proteins that have lost their function because of some damage, but also proteins with changes in structure that have not yet affected their functioning. Moreover, in the case of simultaneous appearance of many proteins with modified structure, the cell commits suicide, undergoing apoptosis or necrosis together with all its proteins, even if most of them have not changed, i.e. remained native.
Age-related weakening of the quality control could save many cells that otherwise would have been destroyed and thus would have exacerbated the reduction of cellularity. Accumulation of cells with random errors in DNA and proteins in the tissues of aging organisms would be a side effect of such a strategy. Gradual weakening of quality control, resulting in the accumulation of errors, is indeed observed in the course of aging; it serves as the main argument for the supporters of aging as the result of random damage. T. Nistrom et al. [30] recently showed that Drosophila aging is accompanied by the reduction of proteasome activity, the key mechanism of protein quality control, and increase in the amount of damaged (carbonylated or oxynonenal-bound) proteins. A similar effect was also described in higher animals (mammals) [31, 32], in humans in vivo [33, 34], and in human cell culture [35]. The concentration of ubiquitin [36] in the enzymes involved in ubiquitin binding to the “victim” protein [37, 38] was shown to decrease with age in certain animal tissues. In addition, inactive mutant forms of ubiquitin preventing “normal” ones from performing their function as protein quality controllers were shown to appear in old animals [39]. However, we should not forget that reduction of cellularity is likely to have been originally programmed in the genome as the final stage of ontogenesis. Thus, we come to the situation when aging, having begun as the result of the relevant program, is gradually turning into the process of accumulation of random (stochastic) damage to biopolymers, which remain unnoticed by the weakened systems of quality control of these polymers [8].
AGING AS A RESULT OF PROGRAMMED OXIDATIVE STRESS
There is much evidence that ROS play the role of the “samurai sword” in the suicide of biological systems of varying complexity. This type of phenomena can be found already in bacteria, where the “toxin–antitoxin” mechanisms in some cases indirectly cause cell death resulting from the sharp rise in ROS level caused by a free toxin [40]. In yeast, the disastrous effect of the pheromone excess ultimately leads to the outbreak of ROS generation in mitochondria [41]. In multicellular organisms, self-destruction of mitochondria can be induced by ROS-mediated opening of pores in the inner mitochondrial membrane [42, 43]. Apoptosis and necrosis of cells of multicellular organisms is accompanied by increase in ROS level and opening of the same mitochondrial pores [42, 43]. Organoptosis (disappearance of an organ in the course of ontogenesis) of the tadpole tail is caused by massive hydrogen peroxide production in the cells of this organ [44]. Age-related increase in ROS level has been described in yeast [45], mycelial fungi [46], plants [47], insects [48], and mammals [49-52]. The lethal effect of abscisic acid generated by seeds of annual plants might also be mediated by reactive oxygen species [53].
In their recent review [51], T. B. Kirkwood and A. Kowald summarize the arguments in favor of a concept according to which aging is causally connected the toxic effect of ROS: 1) ROS are continually formed during respiration in all aerobic cells; 2) ROS can cause oxidative damage to practically all organic compounds; 3) such damage can be observed in organisms and it increases with age; 4) mutations that reduce ROS-caused damages increase the lifespan of the organism; 5) increased lifespan in animals with improved protection against ROS has been shown; this improvement could result from some natural causes or from the introduction of additional genes encoding antioxidant enzymes. In contrast, prooxidants typically reduce lifespan (see below for data on increase in lifespan by small amounts of the prooxidant paraquat).
However, we have recently witnessed an increase in attacks on Harman’s hypothesis on the role of mROS in aging because of the apparent inconsistencies between the accumulating observations and the predictions of its simplest version according to the “pessimists” who believe the age-related increase in ROS danger to be accidental [54]. For example, the naked mole rat forms more ROS than a mouse, and its antioxidant protection is weaker than that of a mouse; therefore, the level of DNA and protein oxidation in higher in these animals. It would seem that the naked mole rats should have shorter lifespan than mice. However, in reality, as we have already noted, they live in the laboratory more than 10 times longer than mice. This could be caused by the fact that the disabling of the aging program in the naked mole rat takes place at some stage after ROS formation (for example, at the level of ROS neutralization by hyaluronan, see above).
The question arises, why did evolution choose ROS as the tool of aging? There is no doubt that ROS, having appeared because of a sharp increase in O2 in the atmosphere around 2.5 billion years ago, are still a problem for modern aerobic life forms. Perhaps that is why aging as a specialized mechanism of evolution is organized so as to foster the improvement of the organism’s antioxidant system. In a sense, ROS act like the fox in the example described above for the improvement of the hare’s breed, but the selection favors not the ability to escape the predator, but the best system of anti-oxygen protection. This circumstance is the direct result of the involvement of ROS in the implementation of the programs of self-destruction of mitochondria, cells, organs, and organisms.
Here it is appropriate to recall also other important functions of ROS, without which the life of modern organisms is no longer possible. The example of tadpoles is quite demonstrative. Their transformation into frogs is accompanied by massive ROS formation causing apoptosis of tail cells, which leads to the disappearance of this organ [44]. However, if a piece of a tail is torn from a young tadpole, its regeneration will require proliferation of cells of the remaining tail part, and this phenomenon will be activated by ROS [55]. Thus, along with the grim function of ROS as the means of suicide of individual cells or the organism, these same compounds can be vital for the same organism under different conditions [8] (for details, see below).
Aging-causing ROS are formed in mitochondria. There are several places of where ROS are formed in cells. Primarily, ROS are generated in mitochondria, the organelles responsible for the absorption of almost all the oxygen entering the organism via its lungs. Each cell has many separate mitochondria. Due to the folding of the inner mitochondrial membrane, its total area is measured in thousands of square meters in the adult human. Over 50% of this membrane is formed by the enzymes catalyzing cellular respiration and ATP synthesis coupled to this process. It is the respiration enzymes that serve as one of the main catalysts of transformation of O2 into superoxide anion-radical, which in turn forms other ROS. Basically, the main function of the mitochondrial respiratory enzymes is to turn O2 into harmless water. But even if only a small part of the oxygen absorbed by mitochondria turns into superoxide, the amount of this potentially poisonous compound will be enough to cause serious problems in our bodies [8].
Thus, mitochondria are potentially the main (or one of the main) ROS generators in mammalian cells. This condition is necessary (but not sufficient) to conclude that it is mitochondrial ROS that cause the slow, growing over years, poisoning of the organism, which we call aging. A number of facts support this view.
1. The lifespans of different species are inversely proportional to ROS production in mitochondria. Bird mitochondria form ROS more slowly than the mitochondria of mammals of the same size whose lifespan is much shorter [14]. Similar relationships are also observed between bats (weight 8 g, live up to 45 years) and shrews (weight 25 g, live 1-2 years) [16, 17]. R. S. Sohal [56], G. Barja [57, 58], and M. Brand [14] have independently shown that the higher is the rate of ROS generation during reverse electron transfer in complex I of the heart mitochondrial respiratory chain, the shorter is the lifespan of the warm-blooded animal. This correlation was not observed when ROS generation was measured for direct electron transfer at the same chain site [14]. The study by the group from Cambridge (UK) was especially thorough: it examined 12 different species of mammals (from mouse and rat to baboon and cow) and birds (quail and pigeon) [14]. The data obtained for 11 species corresponded to a straight line describing the lifespan as the inverse function of the rate of mitochondrial ROS generation. Only one species was an exception from the rule. It was the naked mole rat. However, this is the case when an exception confirms the rule. The fact is that the naked mole rat was the only non-aging being among the studied species. As already mentioned, the aging program in the naked mole rat is apparently blocked at some stage after ROS generation, which explains its discrepancy with the described correlation: ROS are formed, but are neutralized by hyaluronan and cannot transmit the deadly signal further along the chain of events leading to aging.
The outlined facts and considerations suggest that ROS concentration in mitochondria should increase with age in aging organisms. Most recently, this has been directly confirmed (first in drosophila [48], and then in mice with accelerated aging [49]) by Murphy et al. who have developed an elegant method for measuring mitochondrial ROS in vivo using penetrating cations. Significantly, some data have indicated increase in ROS generation on reversal of electron transfer in muscle mitochondria in humans. H2O2 generation by skeletal muscle mitochondria of young people (average age 23.5 years) was shown to be almost three times lower than in older individuals (67.3 years), this effect being blocked by rotenone, an inhibitor of complex I [59].
It is not only the respiratory chain that is responsible for the increased ROS production in mitochondria of aging organisms. For example, the amount of monoamine oxidase A localized in the outer membrane of rat heart mitochondria increases 7.5-fold during its 24 months of life. Monoamine oxidase A catalyzes oxidation of catecholamines, serotonin, and some other amines by oxygen, forming H2O2. This process is of course limited by low concentration of these oxidation substrates, but the scope of the aging effect is so large that it is hardly justified to neglect it completely.
2. Antioxidant protection of mitochondria decreases significantly with age. The decline of the level of the SIRT3 enzyme plays the key role in this effect. SIRT3 is a deacetylase that stimulates the critical mitochondrial antioxidant systems – glutathione reductase and superoxide dismutase 2 [60-63].
3. Reduction of mitochondrial ROS level slows aging. This effect was achieved in three different ways. a) Catalase, the key antioxidant enzyme removing H2O2, was artificially redirected to the mitochondrial matrix (it is normally absent from this compartment). The DNA segment of the catalase gene encoding the 11 amino acid residues at the N-terminus of the protein was removed. This part of the protein forms the address directing it to peroxisomes. Instead of peroxisome address, the gene was complimented with the DNA segment encoding the 25-amino-acid-long polypeptide that forms the mitochondrial address in the ornithine transcarbamylase enzyme. The modified gene was introduced into a mouse embryo. Mice with this mutation were shown to live longer (data obtained in the group of P. Rabinovitch [64]). Studies conducted in the same laboratory have shown that aging of skeletal muscles slows dramatically in the presence of catalase in mitochondria [65]. A number of effects that normally increase with age were no longer present: increased H2O2 production by muscle mitochondria, increased number of mutations in mitochondrial DNA, increase in protein carbonylation (especially that of mitochondrial proteins), and decrease in maximal rate of mitochondrial respiration and coupled ATP synthesis. Age-related reduction of the number of skeletal muscle mitochondria was inhibited [65]. In addition, the adverse aging effect on the myocardium was weakened, the effects of systemic inflammation were decreased, and the frequency of development of tumors of non-hematopoietic origin was reduced; at the same time, catalase had no effect on the development of tumors of the hematopoietic system and glomerulonephropathies [66, 67]. Studies conducted on mice with a mutation in the proofreading domain of mitochondrial DNA-polymerase (“mutator mice”) showed that targeted catalase delivery to heart mitochondria reduces the number of mitochondrial DNA deletions, protein carbonylation, and the amount of the active form of caspase 3, the key enzyme of apoptosis [68]. These beneficial effects were much less pronounced or even absent when catalase had either nuclear or peroxisomal address [64]. b) The gene of protein p66shc was knocked out. The protein forms complexes with mitochondrial cytochrome c and apparently allows this cytochrome to transfer an electron directly to O2, forming O2–.. The lifespan of the mice increased by 30% (data obtained in the group of P. G. Pelicci [69-72]). Interestingly, inactivation of p66shc gene reduced oxidative damage of both mitochondrial and nuclear DNA in vivo in lungs, liver, spleen, skin, skeletal muscles, and kidneys, but it had no effect on this parameter in brain and heart [70]. Such tissue specificity fully corresponds to p66shc content, which is minimal in brain and heart. One can assume that these two organs to a large extent are not affected by the aging program, at least in the part, which is implemented rather early and is mediated by p66shc. c) In our laboratory, we have synthesized a mitochondria-targeted low molecular weight antioxidant (SkQ1). This compound was shown to prolong the lifespan of fungi, plants, crustaceans, insects, fish, and mammals; it also inhibited the development of a large group of age-related diseases and symptoms [6, 8, 73-77].
4. Increased level of mitochondrial ROS accelerates aging. One of the participants of our anti-aging project, the President of the Royal Swedish Academy of Sciences B. Cannon, together with her group conducted experiments on the “mutator” mice. As noted above, the lifespan of these mice is much shorter, and when dying they have all the signs of progeria (premature aging). These effects were strongly attenuated by the addition of the mitochondrial antioxidant SkQ1 to their drinking water.
5. Aging is coupled with the accumulation of errors in mitochondrial DNA [65] and decrease in the amount of the mitochondrial phospholipid cardiolipin [78, 79]. In the above-mentioned progeric mice, especially many mutations are accumulated in the mitochondrial gene of cytochrome b involved in ROS generation in mitochondria. Normally, mutations in mitochondrial DNA occur approximately 10 times faster than in nuclear DNA, despite the presence of a system eliminating damaged mitochondria (mitoptosis), i.e. the process, which should facilitate the cleaning of the cellular population of mitochondria with damaged DNA [4]. This could be explained by damage to mitochondrial DNA due to its oxidation by ROS formed in the inner mitochondrial membrane to which mDNA is attached.
Cardiolipin is the other “weak point” of mitochondria. This unusual phospholipid containing not two, but four fatty acid residues, is found only in the inner mitochondrial membrane, where it is the most common lipid. Most fatty acids (and often all of them) in the cardiolipin molecule are polyunsaturated, i.e. with several double bonds (in all other phospholipids one of the two fatty acids is always saturated). Due to this property, cardiolipin is extremely sensitive to ROS. The cardiolipin dimer is bound to cytochrome b of the respiratory chain. Its eight fatty acids form a sort of well whose walls contain 16 double bonds in case of linoleic acid (the most common fatty acid in cardiolipin). This structure is the perfect “fuse” for “ignition” of the inner mitochondrial membrane: ROS, when attacking this structure, will most likely start a chain reaction of phospholipid peroxidation in the membrane. Cytochrome b, whose active sites are very close to the cardiolipin dimer, can be the source of these ROS [80]. It was shown that increase in the electrical potential difference (Δψ) on the mitochondrial membrane inhibits electron transfer from cytochrome bL heme to bH heme, which in turn results in inability to oxidize the free-radical form of ubiquinone (semiquinone CoQ–.) by cytochrome bL. As a result, CoQ–. accumulates and is then oxidized by molecular oxygen – forming O2–.. Under the same conditions, O2–. is generated also on reverse electron transfer involving complex I. Having been formed in one of these two ways, superoxide attacks cardiolipin, and the reaction product, oxidized cardiolipin, can no longer keep cytochrome c on the outer surface of the inner mitochondrial membrane, and thus cytochrome c is cleaved from the membrane, entering the mitochondrial intermembrane space. Products of cardiolipin oxidation are also released there. The interaction with these products results in cytochrome c gaining cardiolipin-peroxidase activity, which in turn accelerates oxidation of a new portion of cardiolipin and leads to the release of additional cytochrome c from the membrane [81, 82].
Protein p66shc mentioned above is located in the intermembrane space. It also forms complexes with soluble cytochrome c. The complex of cytochrome c and p66shc starts to reduce O2 to O2–., which in turn increases superoxide production by mitochondria.
According to data obtained in our group by M. Y. Vyssokikh, the level of unsaturation of cardiolipin fatty acid residues is reduced in progeric mice with mutant mitochondrial DNA-polymerase (probably as a defense against oxidative stress), and this effect is cancelled by the mitochondria-targeted antioxidant SkQ1.
According to the outlined logics, maximal mammalian lifespan is inversely related to the number of double bonds and the ability for peroxidation of phospholipids in liver mitochondria [83, 84].
Comparison of the queen and worker bees provided another example of the same kind (their lifespans are measured in years in the first case and dozens of days in the second [85]). Worker bees were shown to have much more polyunsaturated fatty acids prone to peroxidation, and queen bees have saturated fatty acids resistant to this risk [86]. The content of cytochrome c per cytochrome oxidase in worker bees is 15 times higher than in the queen bee, which could contribute to cardiolipin peroxidation and O2–. generation by this cytochrome in worker bees [87]. Finally, the amount of antioxidant protein, the juvenile hormone vitellogenin, was much higher in queen bees [87].
6. Mitochondrial ROS cause apoptosis, thereby contributing to the reduction of cellularity of organs and tissues. Extensive experimental material on the involvement of ROS in programmed cell death has already been accumulated. Mitochondrial ROS are particularly important in this respect. Mutant cell lines lacking mitochondrial DNA, and hence the ability for ROS formation in the respiratory chain, are extremely resistant to apoptosis. The mitochondrial antioxidant SkQ1 blocks cell apoptosis in culture caused by the addition of hydrogen peroxide. Added peroxide is rapidly decomposed by cellular enzymes, but after 1-2 h there is a powerful burst of endogenous ROS generation; it is this effect that is neutralized by SkQ1. Here we deal with a phenomenon discovered in our laboratory by D. B. Zorov, namely ROS formation caused by other ROS [88]. SkQ1 was also shown to block cell necrosis in vitro [74]. Furthermore, SkQ1 was found to prevent age-related activation of apoptosis of rat fibroblasts [80].
Thus, there are good reasons to believe that it is mitochondrial ROS that cause the reduction in organ and tissue cellularity in aging organisms [8].
EXAMPLES OF ARTIFICIAL SHUTDOWN OF THE AGING PROGRAM
The programmed aging hypothesis leads to one most important prediction: aging can be canceled by switching off one of the essential steps of the relevant program. Today we already have some examples of this phenomenon.
Plants and invertebrates. Aging of the annual plant Arabidopsis thaliana takes only several weeks and is caused by poisoning of the leaves with compounds formed in the seeds of this plant. Knockout of the two genes required for flowering and fruiting completely prevents rapid aging of arabidopsis and turns it into a perennial, which forms rhizomes and reproduces vegetatively [89, 90].
The lifespan of the short-living nematode C. elegans and of Drosophila may be increased 5-10-fold by mutations of one or two genes encoding proteins of some of the metabolic regulatory cascades (for review see [4]). The female octopus of the species Octopus hummelincki, having jealously guarded her eggs from marine predators, stops eating immediately after the hatching of its young. Her inevitable death from starvation can be prevented by removing the so-called optic glands [91].
Vertebrates. Not only invertebrates, but also vertebrates also provide examples of acute phenoptosis related to reproduction.
Males of an Australian marsupial mouse commit suicide a couple of weeks after the rut, being affected by their own pheromone that was previously used to attract females. This pheromone has receptors in the male vomeronasal organ. Pheromone binding results in the development of a signal, which, in case of prolonged action, somehow blocks the control functions of the hippocampus in relation to the hypothalamus. This results in severe stress due to increased production of corticosteroids, adrenaline, and noradrenaline leading to impaired salt metabolism and acute renal failure. Castration of males or keeping them separately from females increases their lifespan to match that of females [92].
Males of the small South American marsupial Gracilinanus microtarsus die right after mating, and the females die somewhat later, after the end of the lactation period (A. Vercesi, 2013, personal communication). Single breeding species among some amphibians and reptiles and many fish have been described [93]. All the examples listed in this section can be attributed to the acceleration of evolution by the increase in the diversity of the offspring. This diversity will increase if an individual can become a father or mother only once.
Pacific salmon present an example of rapid programmed aging. Before spawning, this fish turns into humpback salmon, a ridiculous humpbacked creature with a mouth not suitable for the intake of food. The humpback salmon dies soon after spawning. Its death can be compared to an accelerated movie where the entire aging program is scrolled during a couple of weeks, starting from decrease in immunity and ending with bone osteoporosis, skeletal muscles sarcopenia, skin thinning, and the development of tumors. Zoologists have long believed that accelerated aging of salmon results from the hard work performed by this fish swimming against the current of the river, sometimes for a thousand kilometers. This hypothesis collapsed when it became clear that this transformation into a humpback salmon could be observed even when the spawning site was only few hundred meters away from the ocean [94]. Apparently, it is the change of seawater for freshwater in the fishes’ habitat that serves as a signal to activate the aging program in salmons. S. Austad suggested that salmon progeria and slow aging of higher vertebrates are of completely different nature: accelerated aging is a program, and slow aging results from the accumulation of errors [95]. However, the above-noted commonality of many signs of the two aging types contradicts this explanation. For example, T. Maldonado et al. [96-98], when studying salmon brain, found the peptides of amyloid plaques, which develop with age in humans with Alzheimer’s disease.
It has been already mentioned that knockout of the gene of protein p66shc slows aging and prolongs the lifespan of mice [69]. Further experiments have shown that the terminal step of the respiratory chain:
can be shunted by the complex of cytochrome c and p66shc:
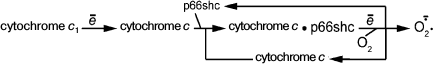 (2)
(2)As a result, superoxide anion is generated instead of harmless water. Knockout of the p66shc gene reduces ROS generation leading to the inhibition of aging [70-72].
In 2014, Blackburn et al. [99] described the reversal of age-related involution of the thymus in yearling mice. They showed that a month-long stimulation of the gene encoding the transcription factor FOXN1 in thymus epithelial cells causes several-fold increase in thymus size and the number of thymocytes, which decrease dramatically in the beginning of the experiment due to aging of the mice. In general, involution of the thymus is an excellent example of the programmed character of immune system aging, and the fact of the reversal of this effect by the stimulation of a single gene directly proves the fundamental possibility of artificial reversal of one of the key signs of mammalian aging.
In 2014, Canaan et al. [100] reported a 20% increase in the median and maximal lifespan in mice with knockout of the gene for protein FAT10. In these mice, the development of a number of signs of aging was dramatically slowed; these signs included sarcopenia, obesity, baldness, graying, and perhaps the most important sign, development of malignant tumors, which were completely absent in the mutants (see also [101-103]). Noncovalent binding of FAT10 with protein MAD2 involved in spindle formation during the cell cycle was previously shown in the same laboratory. This phenomenon caused inhibition of the cycle at the stage of anaphase [104]. Study of the amino acid sequence of FAD10 demonstrated that essentially this protein is a dimer of a somewhat modified ubiquitin, the “black mark” attached to other proteins that should be disassembled to amino acids in proteasomes [105]. It was also shown that FAT10 (1) stimulates ubiquitination of the NFκB protein, the central mediator of innate immunity, and (2) inhibits the expression of the mitochondrial uncoupling proteins UCP1, UCP2, and UCP3, which reduce mROS generation by some decrease in Δψ on the mitochondria membrane [100]. In turn, mROS are known as NFκB activators because they increase the activity of IκBα protein kinase. This kinase phosphorylates IκBα, leading to its subsequent degradation in proteasomes [106].
Aging and cancer as programs of slow and acute phenoptosis. If aging is slow phenoptosis increasing the pressure of natural selection, then cancer can be considered as acute phenoptosis eliminating individuals with increased mutational load [107-110]. It is also possible that cancer limits the lifespan of aging organisms, affecting even those individuals whose mutation load is still low. The latter case would confirm the principle postulated by A. Weismann [111], according to which the reduction in individual lifespan can be a biological adaptation accelerating the change of generations, and hence of evolution.
In this context, the fact that mutation in FAT10 protein not only inhibits the development of age-related symptoms, but it also prevents the development of tumors is of particular importance [100, 103]. FAT10 is apparently involved in both lethal programs (those connected to aging and development of malignant tumors); in this respect, it differs from the mitochondria-targeted antioxidant SkQ1, which inhibits aging and only certain types of cancer [8]. However, SkQ1 requires no intervention in the genetic system of the cell, which reduces the risk of damaging something useful for the organism in the course of our fight against aging.
THE RIDDLE OF PROPERTIES COUNTERPRODUCTIVE TO THE ORGANISM
Among the objections of “pessimistic” gerontologists, there is one that still poses some difficulties for the “optimists” to provide a definitive answer to. This is the question of the possible mechanism of selection of the aging program in the course of evolution, which seems quite difficult to solve, as this program is counterproductive for the individual.
When it comes to acute phenoptosis, there is no doubt that there are many counterproductive programs, which, despite the obvious harm for the organism, have become part of the genomes in the process of biological evolution. These programs include not only death right after breeding, but also rapid aging of many annual plants. In the latter case, genes have been identified that are required for the killing of plant after ripening of its seeds [89, 90]; one of the poisons used as the means of such killing, abscisic acid, has been described [6, 8]. Aging of animals differs from all these phenomena only by its slower pace.
One of the possible explanations of the selection of counterproductive programs is that they are carried out by bifunctional proteins that possess two functions: one harmful for the organism and another extremely useful, disappearance of which would be lethal [8]. For example, cytochrome c is involved in apoptosis induced by ROS, which we believe to play the key role in animal aging. Cytochrome c fulfills this counterproductive function (involvement in the aging process) when it is released from mitochondria to the cytosol to interact with protein Apaf-1. It seems that any mutation inactivating the cytochrome c gene could prevent aging, thereby providing an advantage for the mutant animal in its struggle for survival. However, cytochrome c has also another, “positive” function, electron transfer along the mitochondrial respiratory chain. Disappearance of cytochrome c would cause collapse of mitochondrial respiration similar to poisoning of an animal with cyanide. Interestingly, binding of cytochrome c with Apaf-1 and its respiratory chain partners (cytochrome c1 and cytochrome oxidase) is due to ionic interaction of the “corolla” of lysine cationic groups of cytochrome c with anionic groups of dicarboxylic acids of Apaf-1 [112], cytochrome c1, and cytochrome oxidase [113]. Perhaps it was cytochrome c that was selected in the course of evolution to participate in aging, because this small (only 104 amino acids) single-domain protein was already involved in the vital function of respiration, and a mutation of the corresponding gene affecting the interaction of cytochrome c with Apaf-1 would most likely also lead to the disruption of the respiration. Nevertheless, in our group we have shown that replacement of one of the lysines of the cationic corolla of cytochrome c (K72) by tryptophan leads to the production of a protein fully active with respect to its respiratory function, but it forms an inactive complex with Apaf-1. The K72W mutant was quite viable in vivo (as well as fibroblasts obtained from it [114, 115]). We are now studying the aging of the mutant mice.
An alternative function was also discovered for protein Apaf-1. It was shown to be involved in a cascade of processes providing the arrest of the cell cycle (i.e. cell division) in response to DNA damage. Apaf-1 is transferred from the cytosol to the cell nucleus, where it interacts with checkpoint kinase 1 [116]. Caspases 9 and 3 are the next enzymes in the same apoptotic cascade, where cytochrome c and Apaf-1 are involved. Besides apoptosis, they were found to be essential for the differentiation of stem cells into muscle cells, monocytes, or erythroid cells [117, 118]. Abscisic acid, causing death of annual plants, is a phytohormone regulating a number of vital processes in plants, such as embryonic development, reproduction, cell division and their elongation, protection against stress, etc. So let us not be too quick in qualifying this compound as “lethal poison”. By the way, abscisic acid was found in brain and other mammalian tissues, where its function remains completely unclear [119, 120].
According to J. Mitteldorf [93], aging genes may be largely protected against mutations, similar to the genes encoding the key metabolic enzymes [121-124]. (On uneven accumulation of mutations in different genes, see [125].) The presence of parallel pathways of implementation of programs counterproductive to individual organisms could be another mechanism contributing to their preservation. In such a case, mutation in one of the pathways would not cause complete shutdown of the program [93].
However, recent data on the new function of the above-mentioned apoptotic cascade proteins Apaf-1, caspase 9, and caspase 3 provide perhaps the most meaningful answer to the question of how a counterproductive program could avoid elimination by natural selection. Canadian researchers Hekimi et al. [126] recently showed that inhibition of respiratory chain complexes I or III causes ROS increase in the tissues of the nematode C. elegans. The addition of small (0.1 mM) concentrations of the prooxidant paraquat gives the same result. Amazingly, ROS increase leads to a marked increase in the animals’ lifespan in all these cases. Even more surprisingly, Apaf-1 and caspase 9 were found to be essential to prolong lifespan.
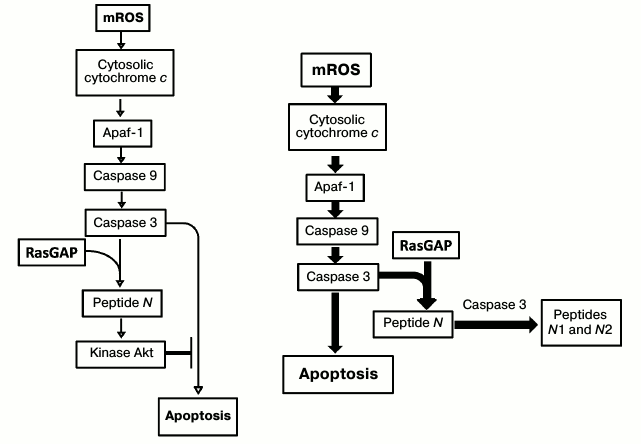
A similar effect was earlier observed in mice by Widmann et al. [118]. In this case, the authors used doxorubicin, which causes death of 50% of rodents after about 50 days after the beginning of intoxication. Absolutely all the mutants without caspase 3 died by this time. Widmann et al. found the mechanism of the saving effect of caspase 3, which was shown to cleave peptide N from the protein p120 RasGAP. Peptide N, in turn, activates the antiapoptotic protein kinase Akt. It is quite remarkable that the affinity of caspase 3 to RasGAP is so high that even small amounts of caspase are sufficient for the emergence of peptide N and activation of Akt. Increased level of caspase 3 leads to cancellation of the order to block apoptosis, because caspase 3 cleaves N peptide into two peptides, N1 and N2, which no longer have the ability to activate Akt. Thus, one type of effect (increase in ROS level) first protects cells against death, and then, with further increase in ROS concentration, provokes death (Fig. 2). It is significant that mechanisms of cell death are different in wild-type mice and caspase 3 mutants. Wild-type cells die of apoptosis, and mutants of necrosis.
Fig. 2. Appearance of small amounts of mROS can inhibit apoptosis, while further increase in mROS leads to its stimulation.
Nystrom et al. [127] found that a metacaspase Mca1-mutant of yeast (Mca1 is a yeast analog of animal caspase) live for a shorter time than the control. Increased metacaspase expression prolonged yeast lifespan, preventing the accumulation of denatured protein aggregates in their cells. The “disaggregase” Hsp104 and proteasomes are involved in this effect. Expression of a cysteine mutant instead of the native caspase caused only partial removal of the effect of lifespan prolongation (cysteine is required for the protease activity of caspase). Thus, metacaspase-mediated lifespan prolongation only partly depended on the caspase function.
So, the obvious harmfulness of the aging program for an individual does not mean that is cannot be selected by evolution. An undeniable fact is that counterproductive programs do exist, and therefore aging may well be one of them [8, 128].
AGING AS A COUNTERPRODUCTIVE PROGRAM THAT CAN BE SACRIFICED IN
CASE OF DETERIORATING LIVING CONDITIONS
In the previous section we discussed the paradoxical result obtained in the group of S. Hekimi [126], namely, the increase in lifespan of C. elegans caused by small amounts of the prooxidant paraquat.
How do we reconcile this result with our concept on the central mediator role of mROS in the aging program?
On one hand, we cannot exclude that C. elegans is an exception to the general rule. This is one of the shortest-living (median lifespan less than 20 days) and smallest (about 1000 cells) organisms among multicellular species. Removal of damaged or “homeless” cells is one of the functions of apoptosis responsible for age-related reduction in organ cellularity. This process is triggered by mitochondrial ROS. According to Hekimi, C. elegans has too few cells, and their lifespan is too short for the justification of the population cleaning from useless or damaged cells in a particular organ [126].
On the other hand, lifespan extension by small ROS doses may also be a general case, since the scheme presented on Fig. 2 can be applied to any organism that has a so-called inner apoptotic cascade triggered by mitochondria. In our experiments with mitochondria-targeted antioxidant, we have never observed a decrease in lifespan caused by small SkQ1 concentrations. In experiments with mammals, fish, insects, crustaceans, plants, and fungi, certain SkQ1 concentrations increased the median lifespan, and decrease in these concentrations gradually reduced the geroprotective effect but never led to its reversal [73]. It is noteworthy that only in one case, namely in C. elegans, SkQ1 had no effect on lifespan (A. P. Grigorenko and E. I. Rogaev, unpublished), which could be due to the absence of mROS-dependent apoptosis in this short-lived organism.
J. Mitteldorf [93] suggested that any effect that seriously complicates the existence of an organism has a chance to prolong its lifespan, within certain limits, due to inhibition of aging. In such a way, an individual tries to compensate the increased energy costs under deteriorating conditions. This explains the effect of small doses of radiation [93]. Here we should also mention the known effect called “hormesis”, when small amounts of some poison (e.g. chloroform [129, 130]) have a positive impact on lifespan.
At first glance, the cases when a slight increase in ROS level prolongs lifespan seem paradoxical. If we deal with mitochondrial ROS, this phenomenon may relate to mitohormesis (a term introduced by M. Ristow et al. [131, 132]). According to these authors, the decrease in outer glucose concentration leads to prolongation of C. elegans lifespan, mediated by ROS increase and the induction of the antioxidant enzyme catalase caused by these ROS [131] (see also Fig. 2). As shown by De Haes et al. [133], the antidiabetic medication metformin has a similar effect. Metformin stimulates respiration of C. elegans, uncoupling it from ATP synthesis. It causes the following chain of events: superoxide production by mitochondria increases; mitochondrial superoxide dismutase turns superoxide into H2O2; hydrogen peroxide, first, increases the level of peroxiredoxin-2, removing H2O2, and second, oxidizes peroxiredoxin-2 forming the dimeric form of this protein. The peroxiredoxin-2 dimer stimulates phosphorylation of protein p38, an activator of transcription factor SKN-1. All these events were found to be necessary for the extension of C. elegans lifespan by metformin [133]. It was also shown that some extension of lifespan could be caused by mild infections [93] or moderate cooling [134] (for details, see M. V. Skulachev et al. in this issue of the journal).
Adverse environmental conditions causing inhibition of the aging program include food restriction and heavy muscular work. The first of these factors serves as a signal of the coming starvation, and the second one imitates the conditions of sudden flight from fire, flood, attack of predators, and similar disasters. No wonder that both food restriction and muscle load prolong the lifespan of a wide variety of organisms and inhibits the development of the same age-related symptoms as the mitochondrial antioxidant SkQ1 (for details see [8]). Weakening of aging organisms seems to be an optional program that can be sacrificed when deterioration of environmental conditions questions the very existence of an individual.
The work was financially supported by the Russian Science Foundation (grant No. 14-24-00107).
REFERENCES
1.Williams, G. C. (1957) Pleiotropy,
natural-selection, and the evolution of senescence, Evolution,
11, 398-411.
2.Skulachev, V. P. (1997) Aging is a specific
biological function rather than the result of a disorder in complex
living systems: biochemical evidence in support of Weismann’s
hypothesis, Biochemistry (Moscow), 62,
1191-1195.
3.Skulachev, V. P. (1999) Phenoptosis: programmed
death of an organism, Biochemistry (Moscow), 64,
1418-1426.
4.Skulachev, V. P. (2003) Aging and programmed death
phenomena, in Topics in Current Genetics, Model Systems in Aging
(Nystrom, T., and Osiewacz, H. D., eds.) Springer Verlag,
Berlin-Heidelberg, pp. 192-237.
5.Skulachev, V. P., and Longo, V. D. (2005) Aging as
a mitochondria-mediated atavistic program: can aging be switched off?
Ann. N. Y. Acad. Sci., 1057, 145-164.
6.Skulachev, V. P. (2012) What is
“phenoptosis” and how to fight it? Biochemistry
(Moscow), 77, 689-706.
7.Libertini, G. (2012) Classification of phenoptotic
phenomena, Biochemistry (Moscow), 77, 707-715.
8.Skulachev, V. P., Skulachev, M. V., and Feniuk, B.
A. (2013) Life without Aging [in Russian], EKSMO, Moscow.
9.Jones, O. R., Scheuerlein, A., Salguero-Gomez, R.,
Camarda, C. G., Schaible, R., Casper, B. B., Dahlgren, J. P., Ehrlen,
J., Garcia, M. B., Menges, E. S., Quintana-Ascencio, P. F., Caswell,
H., Baudisch, A., and Vaupel, J. W. (2014) Diversity of ageing across
the tree of life, Nature, 505, 169-173.
10.Tian, X., Azpurua, J., Hine, C., Vaidya, A.,
Myakishev-Rempel, M., Ablaeva, J., Mao, Z., Nevo, E., Gorbunova, V.,
and Seluanov, A. (2013) High-molecular-mass hyaluronan mediates the
cancer resistance of the naked mole rat, Nature, 499,
346-349.
11.Seluanov, A., Hine, C., Azpurua, J., Feigenson,
M., Bozzella, M., Mao, Z., Catania, K. C., and Gorbunova, V. (2009)
Hypersensitivity to contact inhibition provides a clue to cancer
resistance of naked mole-rat, Proc. Natl. Acad. Sci. USA,
106, 19352-19357.
12.Pletjushkina, O. Y., Fetisova, E. K., Lyamzaev,
K. G., Ivanova, O. Y., Domnina, L. V., Vyssokikh, M. Y., Pustovidko, A.
V., Vasiliev, J. M., Murphy, M. P., Chernyak, B. V., and Skulachev, V.
P. (2005) Long-distance apoptotic killing of cells is mediated by
hydrogen peroxide in a mitochondrial ROS-dependent fashion, Cell
Death Differ., 12, 1442-1444.
13.Pletjushkina, O. Yu., Fetisova, E. K., Lyamzaev,
K. G., Ivanova, O. Yu, Domnina, L. V., Vyssokikh, M. Y., Pustovidko, A.
V., Alekseevski, A. V., Alekseevski, D. A., Vasiliev, J. M., Murphy, M.
P., Chernyak, B. V., and Skulachev, V. P. (2006) Hydrogen peroxide
produced inside mitochondria takes part in cell-to-cell transmission of
apoptotic signal, Biochemistry (Moscow), 71,
60-67.
14.Lambert, A. J., Boysen, H. M., Buckingham, J. A.,
Yang, T., Podlutsky, A., Austad, S. N., Kunz, T. H., Buffenstein, R.,
and Brand, M. D. (2007) Low rates of hydrogen peroxide production by
isolated heart mitochondria associate with long maximum lifespan in
vertebrate homeotherms, Aging Cell, 6, 607-618.
15.Azpurua, J., Ke, Z., Chen, I. X., Zhang, Q.,
Ermolenko, D. N., Zhang, Z. D., Gorbunova, V., and Seluanov, A. (2013)
Naked mole-rat has increased translational fidelity compared with the
mouse, as well as a unique 28S ribosomal RNA cleavage, Proc. Natl.
Acad. Sci. USA, 110, 17350-17355.
16.Brunet-Rossinni, A. K. (2004) Reduced
free-radical production and extreme longevity in the little brown bat
(Myotis lucifugus) versus two non-flying mammals, Mech.
Ageing Dev., 125, 11-20.
17.Brunet-Rossinni, A. K., and Austad, S. N. (2004)
Ageing studies on bats: a review, Biogerontology, 5,
211-222.
18.Sohal, R. S., Ku, H. H., and Agarwal, S. (1993)
Biochemical correlates of longevity in two closely related rodent
species, Biochem. Biophys. Res. Commun., 196, 7-11.
19.Csiszar, A., Labinskyy, N., Zhao, X., Hu, F.,
Serpillon, S., Huang, Z., Ballabh, P., Levy, R. J., Hintze, T. H.,
Wolin, M. S., Austad, S. N., Podlutsky, A., and Ungvari, Z. (2007)
Vascular superoxide and hydrogen peroxide production and oxidative
stress resistance in two closely related rodent species with disparate
longevity, Aging Cell, 6, 783-797.
20.Sohal, R. S., Ferguson, M., Sohal, B. H., and
Forster, M. J. (2009) Life span extension in mice by food restriction
depends on an energy imbalance, J. Nutr., 139,
533-539.
21.Shi, Y., Pulliam, D. A., Liu, Y., Hamilton, R.
T., Jernigan, A. L., Bhattacharya, A., Sloane, L. B., Qi, W.,
Chaudhuri, A., Buffenstein, R., Ungvari, Z., Austad, S. N., and Van
Remmen, H. (2013) Reduced mitochondrial ROS, enhanced antioxidant
defense, and distinct age-related changes in oxidative damage in
muscles of long-lived Peromyscus leucopus, Am. J.
Physiol., 304, R343-355.
22.Csiszar, A., Labinskyy, N., Orosz, Z., Xiangmin,
Z., Buffenstein, R., and Ungvari, Z. (2007) Vascular aging in the
longest-living rodent, the naked mole rat, Am. J. Physiol. Heart
Circ. Physiol., 293, H919-927.
23.Finch, C. E. (1990) Longevity, Senescence and
the Genome, University Chicago Press, Chicago.
24.Finch, C. E. (2009) Update on slow aging and
negligible senescence – a mini-review, Gerontology,
55, 307-313.
25.Delaney, M. A., Nagy, L., Kinsel, M. J., and
Treuting, P. M. (2013) Spontaneous histological lesions of the adult
naked mole rat (Heterocephalus glaber): a retrospective survey
of lesions in a zoo population, Vet. Pathol., 50,
607-621.
26.Lecomte, V. J., Sorci, G., Cornet, S., Jaeger,
A., Faivre, B., Arnoux, E., Gaillard, M., Trouve, C., Besson, D.,
Chastel, O., and Weimerskirch, H. (2010) Patterns of aging in the
long-lived wandering albatross, Proc. Natl. Acad. Sci. USA,
14, 6370-6375.
27.Buffenstein, R. (2005) The naked mole-rat: a new
long-living model for human aging research, J. Gerontol. A. Biol.
Sci. Med. Sci., 60, 1369-1377.
28.Szilard, L. (1959) On the nature of the aging
process, Proc. Natl. Acad. Sci. USA, 45, 30-45.
29.Ciechanover, A. (2012) Intracellular protein
degradation: from a vague idea through the lysosome and the
ubiquitin–proteasome system and onto human diseases and drug
targeting, Neuro-Degenerat. Dis., 10, 7-22.
30.Fredriksson, A., Johansson Krogh, E., Hernebring,
M., Pettersson, E., Javadi, A., Almstedt, A., and Nystrom, T. (2012)
Effects of aging and reproduction on protein quality control in soma
and gametes of Drosophila melanogaster, Aging Cell,
11, 634-643.
31.Nystrom, T. (2005) Role of oxidative
carbonylation in protein quality control and senescence, EMBO
J., 24, 1311-1317.
32.Koga, H., Kaushik, S., and Cuervo, A. M. (2011)
Protein homeostasis and aging: the importance of exquisite quality
control, Ageing Res. Rev., 10, 205-215.
33.Friguet, B., Bulteau, A. L., Chondrogianni, N.,
Conconi, M., and Petropoulos, I. (2000) Protein degradation by the
proteasome and its implications in aging, Ann. N. Y. Acad. Sci.,
908, 143-154.
34.Shringarpure, R., and Davies, K. J. A. (2002)
Protein turnover by the proteasome in aging and disease, Free Rad.
Biol. Med., 32, 1084-1089.
35.Sitte, N., Merker, K., Von Zglinicki, T., Grune,
T., and Davies, K. J. A. (2000) Protein oxidation and degradation
during cellular senescence of human BJ fibroblasts: part I –
effects of proliferative senescence, FASEB J., 14,
2495-2502.
36.Jahngen, J. H., Lipman, R. D., Eisenhauer, D. A.,
Jahngen, E. G. E., and Taylor, A. (1990) Aging and cellular maturation
cause changes in ubiquitin eye lens protein conjugates, Arch.
Biochem. Biophys., 276, 32-37.
37.Ruotolo, R., Grassi, F., Percudani, R., Rivetti,
C., Martorana, D., Maraini, G., and Ottonello, S. (2003) Gene
expression profiling in human age-related nuclear cataract, Mol.
Vision, 9, 538-548.
38.Hawse, J. R., Hejtmancik, J. F., Horwitz, J., and
Kantorow, M. (2004) Identification and functional clustering of global
gene expression differences between age-related cataract and clear
human lenses and aged human lenses, Exp. Eye Res., 79,
935-940.
39.Tsirigotis, M., Zhang, M., Chiu, R. K., Wouters,
B. G., and Gray, D. A. (2001) Sensitivity of mammalian cells expressing
mutant ubiquitin to protein-damaging agents, J. Biol. Chem.,
276, 46073-46078.
40.Engelberg-Kulka, H., Yelin, I., and Kolodkin-Gal,
I. (2009) Activation of a built-in bacterial programmed cell death
system as a novel mechanism of action of some antibiotics, Commun.
Integr. Biol., 2, 211-212.
41.Pozniakovsky, A. I., Knorre, D. A., Markova, O.
V., Hyman, A. A., Skulachev, V. P., and Severin, F. F. (2005) Role of
mitochondria in the pheromone- and amiodarone-induced programmed death
of yeast, J. Cell Biol., 168, 257-269.
42.Skulachev, V. P. (1996) Why are mitochondria
involved in apoptosis? Permeability transition pores and apoptosis as
selective mechanisms to eliminate superoxide-producing mitochondria and
cell, FEBS Lett., 397, 7-10.
43.Skulachev, V. P. (2002) Programmed death
phenomena: from organelle to organism, Ann. N. Y. Acad. Sci.,
959, 214-237.
44.Kashiwagi, A., Hanada, H., Yabuki, M., Kanno, T.,
Ishisaka, R., Sasaki, J., Inoue, M., and Utsumi, K. (1999) Thyroxin
enhancement and the role of reactive oxygen species in tadpole tail
apoptosis, Free Rad. Biol. Med., 26, 1001-1009.
45.Erjavec, N., and Nystrom, T. (2007)
Sir2p-dependent protein segregation gives rise to a superior reactive
oxygen species management in the progeny of Saccharomyces
cerevisiae, Proc. Natl. Acad. Sci. USA, 104,
10877-10881.
46.Osiewacz, H. D. (2003) Aging and mitochondrial
dysfunction in the filamentous fungus Podospora anserina, in
Topics in Current Genetics (Nystrom, T., and Osiewacz, H. D.,
eds.) Springer Verlag, Berlin-Heidelberg, pp. 17-38.
47.Munne-Bosch, S., and Alegre, L. (2002) Plant
aging increases oxidative stress in chloroplasts, Planta,
214, 608-615.
48.Cocheme, H. M., Quin, C., McQuaker, S. J.,
Cabreiro, F., Logan, A., Prime, T. A., Abakumova, I., Patel, J. V.,
Fearnley, I. M., James, A. M., Porteous, C. M., Smith, R. A., Saeed,
S., Carre, J. E., Singer, M., Gems, D., Hartley, R. C., Partridge, L.,
and Murphy, M. P. (2011) Measurement of H2O2
within living Drosophila during aging using a ratiometric mass
spectrometry probe targeted to the mitochondrial matrix, Cell
Metab., 13, 340-350.
49.Logan, A., Shabalina, I. G., Prime, T. A.,
Rogatti, S., Kalinovich, A. V., Hartley, R. C., Budd, R. C., Cannon,
B., and Murphy, M. P. (2014) In vivo levels of mitochondrial
hydrogen peroxide increase with age in mtDNA mutator mice, Aging
Cell, doi: 10.1111/acel.12212.
50.Blagosklonny, M. V. (2008) Aging: ROS or TOR,
Cell Cycle, 7, 3344-3354.
51.Kirkwood, T. B., and Kowald, A. (2012) The
free-radical theory of ageing – older, wiser and still alive:
modeling positional effects of the primary targets of ROS reveals new
support, BioEssays, 34, 692-700.
52.Blagosklonny, M. V. (2013) Aging is not
programmed genetic pseudo-program is a shadow of developmental growth,
Cell Cycle, 12, 3736-3742.
53.Ahlfors, R., Lang, S., Overmyer, K., Jaspers, P.,
Brosche, M., Tauriainen, A., Kollist, H., Tuominen, H., Belles-Boix,
E., Piippo, M., Inze, D., Palva, E. T., and Kangasjarvi, J. (2004)
Arabidopsis radical-induced cell death1 belongs to the WWE
protein–protein interaction domain protein family and modulates
abscisic acid, ethylene, and methyl jasmonate responses, Plant
Cell, 16, 1925-1937.
54.Gladyshev, V. N. (2014) The free radical theory
of aging is dead. Long live the damage theory! Antiox. Redox.
Signal., 20, 727-731.
55.Love, N. R., Chen, Y., Ishibashi, S.,
Kritsiligkou, P., Lea, R., Koh, Y., Gallop, J. L., Dorey, K., and
Amaya, E. (2012) Amputation-induced reactive oxygen species are
required for successful Xenopus tadpole tail regeneration,
Nature Cell Biol., 15, 222-228.
56.Ku, H. H., Brunk, U. T., and Sohal, R. S. (1993)
Relationship between mitochondrial superoxide and hydrogen peroxide
production and longevity of mammalian species, Free Rad. Biol.
Med., 15, 621-627.
57.Barja, G. (1998) Mitochondrial free radical
production and aging in mammals and birds, Ann. N. Y. Acad.
Sci., 854, 224-238.
58.Barja, G., and Herrero, A. (2000) Oxidative
damage to mitochondrial DNA is inversely related to maximum life span
in the heart and brain of mammals, FASEB J., 14,
312-318.
59.Capel, F., Rimbert, V., Lioger, D., Diot, A.,
Rousset, P., Mirand, P. P., Boirie, Y., Morio, B., and Mosoni, L.
(2005) Due to reverse electron transfer, mitochondrial
H2O2 release increases with age in human
vastus lateralis muscle although oxidative capacity is
preserved, Mech. Ageing Dev., 126, 505-511.
60.Qiu, X., Brown, K., Hirschey, M. D., Verdin, E.,
and Chen, D. (2010) Calorie restriction reduces oxidative stress by
SIRT3-mediated SOD2 activation, Cell Metab., 12,
662-667.
61.Someya, S., Yu, W., Hallows, W. C., Xu, J., Vann,
J. M., Leeuwenburgh, C., Tanokura, M., Denu, J. M., and Prolla, T. A.
(2010) Sirt3 mediates reduction of oxidative damage and prevention of
age-related hearing loss under caloric restriction, Cell,
143, 802-812.
62.Tao, R., Coleman, M. C., Pennington, J. D.,
Ozden, O., Park, S. H., Jiang, H., Kim, H. S., Flynn, C. R., Hill, S.,
Hayes McDonald, W., Olivier, A. K., Spitz, D. R., and Gius, D. (2010)
Sirt3-mediated deacetylation of evolutionarily conserved lysine 122
regulates MnSOD activity in response to stress, Mol. Cell,
40, 893-904.
63.Brown, K., Xie, S., Qiu, X., Mohrin, M., Shin,
J., Liu, Y., Zhang, D., Scadden, D. T., and Chen, D. (2013) SIRT3
reverses aging-associated degeneration, Cell Rep., 3,
319-327.
64.Schriner, S. E., Linford, N. J., Martin, G. M.,
Treuting, P., Ogburn, C. E., Emond, M., Coskun, P. E., Ladiges, W.,
Wolf, N., Van Remmen, H., Wallace, D. C., and Rabinovitch, P. S. (2005)
Extension of murine life span by overexpression of catalase targeted to
mitochondria, Science, 308, 1909-1911.
65.Lee, H. Y., Choi, C. S., Birkenfeld, A. L.,
Alves, T. C., Jornayvaz, F. R., Jurczak, M. J., Zhang, D., Woo, D. K.,
Shadel, G. S., Ladiges, W., Rabinovitch, P. S., Santos, J. H.,
Petersen, K. F., Samuel, V. T., and Shulman, G. I. (2010) Targeted
expression of catalase to mitochondria prevents age-associated
reductions in mitochondrial function and insulin resistance, Cell
Metab., 12, 668-674.
66.Dai, D. F., and Rabinovitch, P. S. (2009) Cardiac
aging in mice and humans: the role of mitochondrial oxidative stress,
Trends Cardiovasc. Med., 19, 213-220.
67.Treuting, P. M., Linford, N. J., Knoblaugh, S.
E., Emond, M. J., Morton, J. F., Martin, G. M., Rabinovitch, P. S., and
Ladiges, W. C. (2008) Reduction of age-associated pathology in old mice
by overexpression of catalase in mitochondria, J. Gerontol.
Biol., 63, 813-824.
68.Dai, D. F., Chen, T., Wanagat, J., Laflamme, M.,
Marcinek, D. J., Emond, M. J., Ngo, C. P., Prolla, T. A., and
Rabinovitch, P. S. (2010) Age-dependent cardiomyopathy in mitochondrial
mutator mice is attenuated by overexpression of catalase targeted to
mitochondria, Aging Cell, 9, 536-544.
69.Migliaccio, E., Giorgio, M., Mele, S., Pelicci,
G., Reboldi, P., Pandolfi, P. P., Lanfrancone, L., and Pelicci, P. G.
(1999) The p66shc adaptor protein controls oxidative stress response
and life span in mammals, Nature, 402, 309-313.
70.Trinei, M., Giorgio, M., Cicalese, A., Barozzi,
S., Ventura, A., Migliaccio, E., Milia, E., Padura, I. M., Raker, V.
A., Maccarana, M., Petronilli, V., Minucci, S., Bernardi, P.,
Lanfrancone, L., and Pelicci, P. G. (2002) A p53-p66Shc signaling
pathway controls intracellular redox status, levels of
oxidation-damaged DNA and oxidative stress-induced apoptosis,
Oncogene, 21, 3872-3878.
71.Napoli, C., Martin-Padura, I., de Nigris, F.,
Giorgio, M., Mansueto, G., Somma, P., Condorelli, M., Sica, G., De
Rosa, G., and Pelicci, P. (2003) Deletion of the p66Shc longevity gene
reduces systemic and tissue oxidative stress, vascular cell apoptosis,
and early atherogenesis in mice fed a high-fat diet, Proc. Natl.
Acad. Sci. USA, 100, 2112-2116.
72.Giorgio, M., Migliaccio, E., and Paolucci, D.
(2004) p66shc Is a Signal Transduction Redox Enzyme, 13th
EBEC Meet. Abstr., 27.
73.Anisimov, V. N., Bakeeva, L. E., Egormin, P. A.,
Filenko, O. F., Isakova, E. F., Manskikh, V. N., Mikhelson, V. M.,
Panteleeva, A. A., Pasyukova, E. G., Pilipenko, D. I., Piskunova, T.
S., Popovich, I. G., Roshchina, N. V., Rybina, O. Yu., Saprunova, V.
V., Samoylova, T. A., Semenchenko, A. V., Skulachev, M. V., Spivak, I.
M., Tsybul’ko, E. A., Tyndyk, M. L., Vyssokikh, M. Y., Yurova, M.
N., Zabezhinsky, M. A., and Skulachev, V. P. (2008)
Mitochondria-targeted plastoquinone derivatives as tools to interrupt
execution of the aging program. 5. SkQ1 prolongs lifespan and prevents
development of traits of senescence, Biochemistry
(Moscow), 73, 1329-1342.
74.Skulachev, V. P., Anisimov, V. N., Antonenko, Y.
N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., Filenko, O. F.,
Kalinina, N. I., Kapelko, V. I., Kolosova, N. G., Kopnin, B. P.,
Korshunova, G. A., Lichinitser, M. R., Obukhova, L. A., Pasyukova, E.
G., Pisarenko, O. I., Roginsky, V. A., Ruuge, E. K., Senin, I. I.,
Severina, I. I., Skulachev, M. V., Spivak, I. M., Tashlitsky, V. N.,
Tkachuk, V. A., Vyssokikh, M. Y., Yaguzhinsky, L. S., and Zorov, D. B.
(2009) An attempt to prevent senescence: a mitochondrial approach,
Biochim. Biophys. Acta, 1787, 437-461.
75.Skulachev, M. V., Antonenko, Y. N., Anisimov, V.
N., Chernyak, B. V., Cherepanov, D. A., Chistyakov, V. A., Egorov, M.
V., Kolosova, N. G., Korshunova, G. A., Lyamzaev, K. G., Plotnikov, E.
Y., Roginsky, V. A., Savchenko, A. Y., Severina, I. I., Severin, F. F.,
Shkurat, T. P., Tashlitsky, V. N., Shidlovsky, K. M., Vyssokikh, M. Y.,
Zamyatnin, A. A., Jr., Zorov, D. B., and Skulachev, V. P. (2011)
Mitochondrial-targeted plastoquinone derivatives. Effect on senescence
and acute age-related pathologies, Curr. Drug Targets,
12, 800-826.
76.Anisimov, V. N., Egorov, M. V., Krasilshchikova,
M. S., Lyamzaev, K. G., Manskikh, V. N., Moshkin, M. P., Novikov, E.
A., Popovich, I. G., Rogovin, K. A., Shabalina, I. G., Shekarova, O.
N., Skulachev, M. V., Titova, T. V., Vygodin, V. A., Vyssokikh, M. Y.,
Yurova, M. N., Zabezhinsky, M. A., and Skulachev, V. P. (2011) Effects
of the mitochondria-targeted antioxidant SkQ1 on lifespan of rodents,
Aging (Albany NY), 3, 1110-1119.
77.Skulachev, V. P. (2013) Cationic antioxidants as
a powerful tool against mitochondrial oxidative stress, Biochem.
Biophys. Res. Commun., 441, 275-279.
78.Petrosillo, G., Matera, M., Casanova, G.,
Ruggiero, F. M., and Paradies, G. (2008) Mitochondrial dysfunction in
rat brain with aging. Involvement of complex I, reactive oxygen species
and cardiolipin, Neurochem. Int., 53, 126-131.
79.Paradies, G., Petrosillo, G., Paradies, V., and
Ruggiero, F. M. (2010) Oxidative stress, mitochondrial bioenergetics,
and cardiolipin in aging, Free Rad. Biol. Med., 48,
1286-1295.
80.Skulachev, V. P., Antonenko, Y. N., Cherepanov,
D. A., Chernyak, B. V., Izyumov, D. S., Khailova, L. S., Klishin, S.
S., Korshunova, G. A., Lyamzaev, K. G., Pletjushkina, O. Y., Roginsky,
V. A., Rokitskaya, T. I., Severin, F. F., Severina, I. I., Simonyan, R.
A., Skulachev, M. V., Sumbatyan, N. V., Sukhanova, E. I., Tashlitsky,
V. N., Trendeleva, T. A., Vyssokikh, M. Y., and Zvyagilskaya, R. A.
(2010) Prevention of cardiolipin oxidation and fatty acid cycling as
two antioxidant mechanisms of cationic derivatives of plastoquinone
(SkQs), Biochim. Biophys. Acta, 1797, 878-889.
81.Kagan, V. E., Borisenko, G. G., Tyurina, Y. Y.,
Tyurin, V. A., Jiang, J. F., Potapovich, A. I., Kini, V., Amoscato, A.
A., and Fujii, Y. (2004) Oxidative lipidomics of apoptosis: redox
catalytic interactions of cytochrome c with cardiolipin and
phosphatidylserine, Free Rad. Biol. Med., 37,
1963-1985.
82.Kagan, V. E., Tyurin, V. A., Jiang, J. F.,
Tyurina, Y. Y., Ritov, V. B., Amoscato, A. A., Osipov, A. N., Belikova,
N. A., Kapralov, A. A., Kini, V., Vlasova, I. I., Zhao, Q., Zou, M. M.,
Di, P., Svistunenko, D. A., Kurnikov, I. V., and Borisenko, G. G.
(2005) Cytochrome c acts as a cardiolipin oxygenase required for
release of proapoptotic factors, Nature Chem. Biol., 1,
223-232.
83.Pamplona, R., Portero-Otin, M., Riba, D., Ruiz,
C., Prat, J., Bellmunt, M. J., and Barja, G. (1998) Mitochondrial
membrane peroxidizability index is inversely related to maximum life
span in mammals, J. Lipid Res., 39, 1989-1994.
84.Barja, G. (2013) Updating the mitochondrial free
radical theory of aging: an integrated view, key aspects, and
confounding concepts, Antioxid. Redox. Signal., 19,
1420-1445.
85.Remolina, S. C., and Hughes, K. A. (2008)
Evolution and mechanisms of long life and high fertility in queen honey
bees, Age (Dordr.), 30, 177-185.
86.Haddad, L. S., Kelbert, L., and Hulbert, A. J.
(2007) Extended longevity of queen honey bees compared to workers is
associated with peroxidation-resistant membranes, Exp.
Gerontol., 42, 601-609.
87.Corona, M., Hughes, K. A., Weaver, D. B., and
Robinson, G. E. (2005) Gene expression patterns associated with queen
honeybee longevity, Mech. Ageing Dev., 126,
1230-1238.
88.Zorov, D. B., Filburn, C. R., Klotz, L. O.,
Zweier, J. L., and Sollott, S. J. (2000) Reactive oxygen species
(ROS)-induced ROS release: a new phenomenon accompanying induction of
the mitochondrial permeability transition in cardiac myocytes, J.
Exp. Med., 192, 1001-1014.
89.Melzer, S., Lens, F., Gennen, J., Vanneste, S.,
Rohde, A., and Beeckman, T. (2008) Flowering-time genes modulate
meristem determinacy and growth form in Arabidopsis thaliana,
Nat. Genet., 40, 1489-1492.
90.Lens, F., Smets, E., and Melzer, S. (2012) Stem
anatomy supports Arabidopsis thaliana as a model for insular
woodiness, New Phytol., 193, 12-17.
91.Wodinsky, J. (1977) Hormonal inhibition of
feeding and death in octopus: control by optic gland secretion,
Science, 198, 948-951.
92.Bradley, A. J., McDonald, I. R., and Lee, A. K.
(1980) Stress and mortality in a small marsupial (Antechinus
stuartii Macleay), Gen. Comp. Endocrinol., 40,
188-200.
93.Mitteldorf, J., and Sagan, D. (2014) Suicide
Genes, MacMillan Press, in press.
94.Skulachev, V. P. (2005) Ageing as atavistic
program which can be cancelled, Vestnik RAN, 75,
831-843.
95.Austad, S. N. (1997) Why We Age? John
Willey & Sons, New York.
96.Maldonado, T. A., Jones, R. E., and Norris, D. O.
(2000) Distribution of beta-amyloid and amyloid precursor protein in
the brain of spawning (senescent) salmon: a natural, brain-aging model,
Brain Res., 858, 237-251.
97.Maldonado, T. A., Jones, R. E., and Norris, D. O.
(2002) Intraneuronal amyloid precursor protein (APP) and appearance of
extracellular beta-amyloid peptide (abeta) in the brain of aging
kokanee salmon, J. Neurobiol., 53, 11-20.
98.Maldonado, T. A., Jones, R. E., and Norris, D. O.
(2002) Timing of neurodegeneration and beta-amyloid (Abeta) peptide
deposition in the brain of aging kokanee salmon, J. Neurobiol.,
53, 21-35.
99.Bredenkamp, N., Nowell, C. S., and Blackburn, C.
C. (2014) Regeneration of the aged thymus by a single transcription
factor, Development, 141, 1627-1637.
100.Canaan, A., DeFuria, J., Perelman, E., Schultz,
V., Seay, M., Tuck, D., Flavell, R. A., Snyder, M. P., Obin, M. S., and
Weissman, S. M. (2014) Extended lifespan and reduced adiposity in mice
lacking the FAT10 gene, Proc. Natl. Acad. Sci. USA, 111,
5313-5318.
101.Ren, J., Wang, Y., Gao, Y., Mehta, S. B., and
Lee, C. G. (2011) FAT10 mediates the effect of TNF-alpha in inducing
chromosomal instability, J. Cell Sci., 124,
3665-3675.
102.Merbl, Y., Refour, P., Patel, H., Springer, M.,
and Kirschner, M. W. (2013) Profiling of ubiquitin-like modifications
reveals features of mitotic control, Cell, 152,
1160-1172.
103.Gao, Y., Theng, S. S., Zhuo, J., Teo, W. B.,
Ren, J., and Lee, C. G. (2014) FAT10, an ubiquitin-like protein,
confers malignant properties in non-tumorigenic and tumorigenic cells,
Carcinogenesis, 35, 923-934.
104.Liu, Y. C., Pan, J., Zhang, C., Fan, W.,
Collinge, M., Bender, J. R., and Weissman, S. M. (1999) A MHC-encoded
ubiquitin-like protein (FAT10) binds noncovalently to the spindle
assembly checkpoint protein MAD2, Proc. Natl. Acad. Sci. USA,
96, 4313-4318.
105.Hipp, M. S. (2005) NUB1L and FAT10, two
ubiquitin-like proteins involved in protein degradation: Thesis,
Universitaet Konstanz.
106.Maryanovich, M., and Gross, A. (2013) A ROS
rheostat for cell fate regulation, Trends Cell. Biol.,
23, 129-134.
107.Sommer, S. S. (1994) Does cancer kill the
individual and save the species? Human Mutation, 3,
166-169.
108.Manskikh, V. N. (2004) Essays on
Evolutionary Oncology [in Russian], SibGMU, Tomsk.
109.Manskikh, V. N. (2008) Hypothesis: phagocytosis
of aberrant cells protects long-loved vertebrate species from tumors,
Uspekhi Gerontol., 21, 27-33.
110.Lichtenstein, A. V. (2005) Cancer as a
programmed death of an organism, Biochemistry (Moscow),
70, 1055-1064.
111.Weismann, A. (1989) Essays upon Heredity and
Kindred Biological Problems, Calderon Press, Oxford.
112.Yu, T., Wang, X., Purring-Koch, C., Wei, Y.,
and McLendon, G. L. (2001) A mutational epitope for cytochrome c
binding to the apoptosis protease activation factor-1, J. Biol.
Chem., 20, 13034-13038.
113.Skulachev, V. P., Bogachev, A. V., and
Kasparinsky, F. O. (2013) Principles of Bioenergetics, Springer
Verlag, Berlin-Heidelberg.
114.Sharonov, G. V., Feofanov, A. V., Bocharova, O.
V., Astapova, M. V., Dedukhova, V. I., Chernyak, B. V., Dolgikh, D. A.,
Arseniev, A. S., Skulachev, V. P., and Kirpichnikov, M. P. (2005)
Comparative analysis of proapoptotic activity of cytochrome c
mutants in living cells, Apoptosis, 10, 797-808.
115.Mufazalov, I. A., Penkov, D. N., Chernyak, B.
V., Pletyushkina, O. Yu., Vyssokikh, M. Y., Chertkova, R. V.,
Kirpichnikov, M. P., Dolgikh, D. A., Kruglov, A. A., Kuprash, D. V.,
Skulachev, V. P., and Nedospasov, S. A. (2009) Preparation and
characterization of mouse embryonic fibroblasts with K72W mutation in
somatic cytochrome c gene, Mol. Biol., 43,
596-603.
116.Zermati, Y., Mouhamad, S., Stergiou, L., Besse,
B., Galluzzi, L., Boehrer, S., Pauleau, A. L., Rosselli, F.,
D’Amelio, M., Amendola, R., Castedo, M., Hengartner, M., Soria,
J. C., Cecconi, F., and Kroemer, G. (2007) Nonapoptotic role for Apaf-1
in the DNA damage checkpoint, Mol. Cell, 28, 624-637.
117.Murray, T. V., McMahon, J. M., Howley, B. A.,
Stanley, A., Ritter, T., Mohr, A., Zwacka, R., and Fearnhead, H. O.
(2008) A non-apoptotic role for caspase-9 in muscle differentiation,
J. Cell Sci., 121, 3786-3793.
118.Khalil, H., Peltzer, N., Walicki, J., Yang, J.
Y., Dubuis, G., Gardiol, N., Held, W., Bigliardi, P., Marsland, B.,
Liaudet, L., and Widmann, C. (2012) Caspase-3 protects stressed organs
against cell death, Mol. Cell Biol., 32, 4523-4533.
119.Le Page-Degivry, M. T., Bidard, J. N., Rouvier,
E., Bulard, C., and Lazdunski, M. (1986) Presence of abscisic acid, a
phytohormone, in the mammalian brain, Proc. Natl. Acad. Sci.
USA, 83, 1155-1158.
120.Bruzzone, S., Basile, G., Mannino, E., Sturla,
L., Magnone, M., Grozio, A., Salis, A., Fresia, C., Vigliarolo, T.,
Guida, L., De Flora, A., Tossi, V., Cassia, R., Lamattina, L., and
Zocchi, E. (2012) Autocrine abscisic acid mediates the UV-B-induced
inflammatory response in human granulocytes and keratinocytes, J.
Cell. Physiol., 227, 2502-2510.
121.Wolfe, K. H., Sharp, P. M., and Li, W. H.
(1989) Mutation rates differ among regions of the mammalian genome,
Nature, 337, 283-285.
122.Sniegowski, P. D., Gerrish, P. J., and Lenski,
R. E. (1997) Evolution of high mutation rates in experimental
populations of E. coli, Nature, 387, 703-705.
123.Hempenstall, S., Picchio, L., Mitchell, S. E.,
Speakman, J. R., and Selman, C. (2010) The impact of acute caloric
restriction on the metabolic phenotype in male C57BL/6 and DBA/2 mice,
Mech. Ageing Dev., 131, 111-118.
124.Sun, H., Skogerbo, G., Wang, Z., Liu, W., and
Li, Y. X. (2008) Structural relationships between highly conserved
elements and genes in vertebrate genomes, PLoS One, 3,
e3727.
125.Wright, B. E. (2004) Stress-directed adaptive
mutations and evolution, Mol. Microbiol., 52,
643-650.
126.Yee, C., Yang, W., and Hekimi, S. (2014) The
intrinsic apoptosis pathway mediates the pro-longevity response to
mitochondrial ROS in C. elegans, Cell, 157,
897-909.
127.Hill, S. M., Hao, X., Liu, B., and Nystrom, T.
(2014) Life-span extension by a metacaspase in the yeast
Saccharomyces cerevisiae, Science, doi:
10.1126/science.1252634.
128.Skulachev, V. P. (2011) SkQ1 treatment and food
restriction – two ways to retard an aging program of organisms,
Aging (Albany), 3, 1045-1050.
129.Heywood, R., Sortwell, R. J., Noel, P. R. B.,
Street, A. E., Prentice, D. E., Roe, F. J. C., Wadsworth, P. F.,
Worden, A. N., and Vanabbe, N. J. (1979) Safety evaluation of
toothpaste containing chloroform. 3. Long-term study in beagle dogs,
J. Environ. Pathol. Tox., 2, 835-851.
130.Palmer, A. K., Street, A. E., Roe, F. J. C.,
Worden, A. N., and Vanabbe, N. J. (1979) Safety evaluation of
toothpaste containing chloroform. 2. Long-term studies in rats, J.
Environ. Pathol. Tox., 2, 821-833.
131.Schulz, T. J., Zarse, K., Voigt, A., Urban, N.,
Birringer, M., and Ristow, M. (2007) Glucose restriction extends
Caenorhabditis elegans life span by inducing mitochondrial
respiration and increasing oxidative stress, Cell Metab.,
6, 280-293.
132.Ristow, M., and Schmeisser, K. (2014)
Mitohormesis: promoting health and lifespan by increased levels of
reactive oxygen species (ROS), Dose Response, 12,
288-341.
133.De Haes, W., Frooninckx, L., Van Assche, R.,
Smolders, A., Depuydt, G., Billen, J., Braeckman, B. P., Schoofs, L.,
and Temmerman, L. (2014) Metformin promotes lifespan through
mitohormesis via the peroxiredoxin PRDX-2, Proc. Natl. Acad. Sci.
USA, doi: 10.1073/pnas.1321776111.
134.Xiao, R., Zhang, B., Dong, Y. M., Gong, J. K.,
Xu, T., Liu, J. F., and Xu, X. Z. S. (2013) A genetic program promotes
C. elegans longevity at cold temperatures via a thermosensitive
TRP channel, Cell, 152, 806-817.