REVIEW: The Programmed Aging Paradigm: How We Get Old
Giacinto Libertini
Independent Researcher; E-mail: giacinto.libertini@tin.itBiochemistry (Moscow) Office, Maronovsky Pereulok 26, 119049 Moscow, Russia; fax: (499) 238-2479; E-mail: biochem@naukaran.ru; ozrina@bio.chem.msu.ru
Received April 28, 2014
According to the traditional explanations (“old paradigm”), aging is due to the progressive accumulation of heterogeneous damages that are insufficiently contrasted by natural selection. An opposite interpretation (“new paradigm”) sees aging as selectively advantageous in terms of supra-individual natural selection, and this implies the indispensable existence of genetically controlled specific mechanisms that determine it. The aim of this work is to expound synthetically the progressive alterations that mark the aging by showing how these changes are clearly defined and regulated by genes. The possibility of such a description, based on sound evidence, is an essential element for the plausibility of the new paradigm, and a fundamental argument against the tenability of the old paradigm.
KEY WORDS: aging, phenoptosis, programmed aging paradigm, non-programmed aging paradigm, cell turnover, cell senescence, Alzheimer, ARMDDOI: 10.1134/S0006297914100034
For “aging”, here precisely defined as “age-related progressive fitness decline/mortality increase”, there are two antithetical general explanations [1] that have very important opposed implications and, so, deserve the definition of paradigms.
The first, here defined as “old paradigm”, explains aging as the effect of various damaging factors insufficiently opposed by natural selection [2]. This paradigm implies that natural selection is successful in the achievement of numberless extraordinary functions and organs, while for contrasting aging it is capable only of limited effectiveness!
The second explanation, here defined as “new paradigm”, justifies aging as a physiologic phenomenon determined and favored, in particular conditions, by supra-individual selection [3]. This paradigm implies that natural selection is successful in the achievement of aging as in other numberless extraordinary functions and organs!
The two paradigms, by definition, are alternative and incompatible with each other.
For the new paradigm, aging is a particular type of “phenoptosis”, an important neologism proposed by Skulachev [4, 5], which includes a large category of well-known phenomena [6] characterized by the self-sacrifice of an individual, genetically caused/induced and regulated, and favored by natural selection, clearly in terms of supra-individual selection. Examples of phenoptosis types, well described by Finch [6], are as follows: aphagy, autogeny, hormonally triggered senescence in plants, death after spawning, death of the male associated with mating/reproduction, endotoxic matricide, ..., and, according to the new paradigm, – aging (“slow phenoptosis” [7]).
Here, I do not want to discuss arguments and evidence for or against the two paradigms, but only focus on a key topic: how we age, i.e. a general description of the aging process in our species (as for mammals in general) on the basis of mechanisms genetically determined and regulated.
In fact, the new paradigm predicts and requires the existence of specific mechanisms, genetically determined and regulated, which cause aging [8]. On the contrary, the old paradigm excludes the possibility that such mechanisms exist: their existence would therefore demonstrate that the old paradigm is false [2].
Only manifest and accepted evidence will be used in the following exposition.
EVIDENCE
Programmed cell death. Cells may die by necrosis, as a result of accidental events (infection, mechanical stress, trauma, ischemia, etc.), or by one of various types of programmed cell death (PCD), e.g. (i) keratinization of epidermis or hair cells; (ii) detachment of cells from the lining of intestines or other body cavities; (iii) osteocytes phagocytized by osteoclasts; (iv) transformation of erythroblasts in erythrocytes and their subsequent removal by macrophages; (v) apoptosis, an ordinate process of self-destruction with non-damaging disposal of cellular debris that makes it different from necrosis. The phenomenon was for the first time described and clearly differentiated from necrosis in the observation of normal liver hepatocytes [9]. A pivotal function of apoptosis in vertebrates is related to the cell turnover in healthy adult organs, as well documented for many tissues and organs [10]. It must be underlined that the term PCD is often used as synonymous of apoptosis, but this is a wrong simplification!
Cell turnover. The endless death of cells by PCD is balanced by an equal proliferation of appropriate stem cells: “Each day, approximately 50 to 70 billion cells perish in the average adult because of programmed cell death (PCD). Cell death in self-renewing tissues, such as the skin, gut, and bone marrow, is necessary to make room for the billions of new cells produced daily. So massive is the flux of cells through our bodies that, in a typical year, each of us will produce and, in parallel, eradicate, a mass of cells equal to almost our entire body weight” [11]. This “massive” turnover (about 690,000 cells per second!) is restricted by duplication limits caused by the telomere–telomerase system (see below).
Cell turnover is a general pattern in vertebrates, but not for all animals (e.g. the adult stage of the worm Caenorhabditis elegans has a fixed number of cells) [6].
The rhythm of cell turnover varies greatly depending on cell type and organ, e.g. in the intestinal epithelium “cells are replaced every three to six days” [12], while “bone has a turnover time of about ten years in humans” [12] and “the heart is replaced roughly every 4.5 years” [13]. Other data about cell turnover rhythms are reported elsewhere [14].
Limits in cell duplication. Cell replication, which is essential for cell turnover, is restricted by known mechanisms. Limits in the number of cell duplication were demonstrated by Hayflick in 1961 [15]. Olovnikov hypothesized that, as DNA molecule shortens at each duplication, this could explain the finite number of duplications [16]. (In fact, it was later documented, for many cell types, that telomere length shows an age-related progressive shortening [17].) The end of DNA molecule (telomere) was demonstrated, first in a protozoan species, to be a simple repeated sequence of nucleotides [18]. The discovery of telomerase, which added other sequences of the nucleotides, was a necessary explanation for cells, as those of germ line, capable of numberless divisions [19]. Telomerase was shown to be repressed by regulatory proteins [20].
In cells where telomerase is not active, an infinite number of duplications is impossible for the progressive shortening of the telomere. Before telomeres reach their minimum length, two phenomena are described.
1) “On/off” cell senescence. In a cell in “cycling” state, the telomere, whatever its length, oscillates between two phases: “capped” and “uncapped” (by a protein complex). The probability of the uncapped phase is inversely proportional to the relative reduction of telomere length. In the uncapped phase, the cell is vulnerable to the transition to non-cycling state, i.e. to the activation of cell senescence program [21] (Fig. 1).
Fig. 1. “On/off” cell senescence. Telomere (DNA end part marked by dots) oscillates between two possible states: uncapped or capped by a proteinic hood. As telomere progressively shortens, the probability of being in the uncapped state increases and in this state the chromosome is vulnerable to homologous recombination and so to cellular senescence [21].
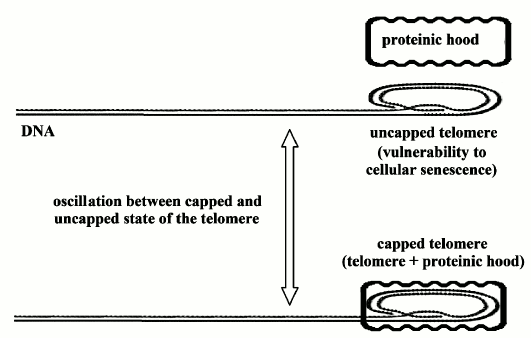
Cell senescence, which can also be activated by other factors, is determined by a mechanism in which the p53 protein is involved and is characterized by the block of the cell cycle besides a long series of changes in the expression of cellular genes. These changes also include modifications of cellular secretions that cause alterations of the extracellular matrix, inflammation, reduced secretion of important structural proteins such as elastin and collagen, and impairments of the surrounding cells [22].
Cell senescence with its stereotyped and predictable alterations has been described as a “fundamental cellular program” [23].
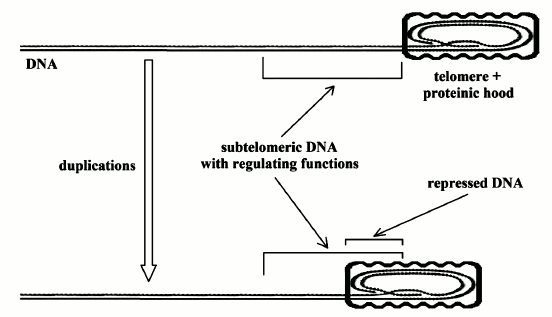
2) “Gradual” cell senescence. The progressive shortening of the telomeres has another effect. The telomere is covered (capped) by a protein complex that, as the telomere shortens, hides the subtelomeric DNA and causes transcriptional silencing (Fig. 2).
“As the telomere shortens, the hood slides further down the chromosome... The result is an alteration of transcription from portions of the chromosome immediately adjacent to the telomeric complex, usually causing transcriptional silencing, although the control is doubtless more complex than merely telomere effect through propinquity… These silenced genes may in turn modulate other, more distant genes (or set of genes). There is some direct evidence for such modulation in the subtelomere...” [22].
Fig. 2. “Gradual” cell senescence. Telomere (DNA end part marked by dots) is capped by a proteinic hood. As telomere progressively shortens, an increasing part of subtelomeric DNA is also capped by the proteinic hood. It is likely that the subtelomeric DNA has regulating functions and its progressive capping alters this regulation and so the expression of many genes [22].
These phenomena (“on/off” and “gradual” cell senescence) progressively affect the mean functioning of the cells in a tissue and the intercellular environment. But, by the activation of telomerase, cell senescence and all related alterations are completely cancelled [24-28].
“Telomerase gene transfection (“telomerization”) is an experimental determinant, switching somatic cells from mortal to immortal without disruption of the remainder of gene expression… This process of gene control is central to cell aging and experimental intervention. Resetting gene expression occurs in knockout mice, cloning, and other interventions, permitting us to make sense of how cell senescence causes aging in organisms... Cells do not senesce because of wear and tear, but because they permit wear and tear to occur because of an altered gene expression. Telomerization effectively replaces the score, allowing the gene to express their previous pattern… Cells do not senesce because they are damaged, but permit damage because they senesce. Homeostatic processes suffice indefinitely in germ cell lines; they suffice in somatic cells if senescence is abrogated” [22].
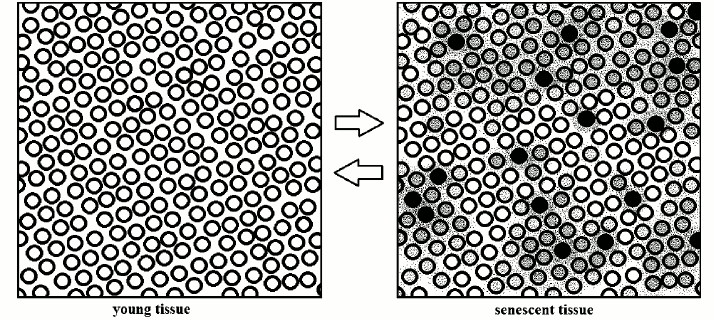
With the passage of time (and with very different rhythms, varying for cell types and organs), in a tissue there is an increase of the percentage of cells: (i) in senescent state; (ii) having functions more or less affected by the shortening of telomeres and the consequent interference in the subtelomeric region; (iii) affected by altered secretions of other cells (Fig. 3).
Fig. 3. Schematic interpretation of the transformation of a young tissue into a senescent tissue: “a modicum of cells displays varying degrees of senescent change” [22]. The inverse transformation is demonstrated: in vitro, for single cells [24-28] and, in vivo, in aged telomerase-deficient mice [29].
This leads, for each tissue and organ to the “atrophic syndrome”, which is characterized by [10]: 1) reduced mean cell duplication capacity and slackened cell turnover; 2) reduced number of cells (atrophy); 3) substitution of missing specific cells with nonspecific cells; 4) hypertrophy of the remaining specific cells; 5) altered functions of cells with shortened telomeres or definitively in noncycling state; 6) alterations of the surrounding milieu and of the cells depending on the functionality of the senescent or missing cells; 7) vulnerability to cancer because of dysfunctional telomere-induced instability [30].
AGING IN OUR SPECIES
The simple concepts outlined in the previous section permit an easy concise description of what characterizes aging. For brevity, this description – already, in part, expounded elsewhere [10] – will be outlined here only for some types of cells and tissues.
Endothelium. The right functionality of endothelium is fundamental to avoid atherogenesis and its complications. The turnover of endothelial cells is assured by endothelial progenitor cells (EPCs), which derive from bone marrow. EPC number is inversely related to age, reduced by cardiovascular risk factors (cigarette smoking, diabetes, hypertension, hypercholesterolemia, etc.), and increased by drugs, such as statins, which protect organ integrity [31].
A slackened turnover of endothelial cells increases the probability of endothelial dysfunction and, therefore, of diseases derived from altered blood circulation (cerebral ischemia, cardiac infarctions, and other diseases caused by compromised blood circulation). Moreover, with negative relation, the number of EPCs is a predictor of cardiovascular risk equal to or more significant than Framingham risk score [31, 32].
In the senile state, diseases deriving from a compromised endothelial function increase exponentially in correlation with age, even if other cardiovascular risk factors are absent [33]. These factors anticipate and amplify the age-related risk [33], while drugs with organ protection qualities, such as statins [34], ACE-inhibitors, and sartans [35], counter their effects.
Heart. An old and deep-rooted belief is that the heart is an organ incapable of regeneration and without cell turnover. On the contrary, “the heart is a self-renewing organ” [13]: in a normal heart, every day about 3 million myocytes die by apoptosis and are replaced by cardiac stem cells: “the entire cell population of the heart is replaced approximately every 4.5 years… The human heart replaces completely its myocyte population about 18 times during the course of life, independently from cardiac diseases” [13].
Cardiac stem cells duplicate and differentiate, allowing myocyte turnover, and show age-related telomeric shortening and cell senescence [36-38]. In the old heart, there is a global loss of myocytes, with a progressive increase in myocyte cell volume per nucleus [39]. The decreasing number of myocytes is due the progressive decline in the ability to duplication of cardiac stem cells [13].
The decline of cardiac contractile capacities causes an enlargement of the heart that conceals the underlying atrophy of the contractile cells. So, in apparent contradiction, the heart chambers are dilated and the senile heart, although atrophic as number of cells, is morphologically hypertrophic [40].
“With aging, there is also a progressive reduction in the number of pacemaker cells in the sinus node, with 10% of the number of cells present at age 20 remaining at age 75... Age-associated left ventricular hypertrophy is caused by an increase in the volume but not in the number of cardiac myocytes. Fibroblasts undergo hyperplasia, and collagen is deposited in the myocardial interstitium” [40].
The heart shows “...some increase in the amount of fibrous tissue and fat in the atrial myocardium with a decrease in the number of muscle fibers, and loss of fibers in the bifurcating main bundle of His and at the junction of the main bundle and its left fascicles, with lesser degrees of loss in the distal bundle branches” [41].
Drugs effective in “organ protection”, as ACE-inhibitors, sartans, and statins, are effective in the prevention of atrial fibrillation [42, 43].
Skin. “Stratum corneal thickness is unchanged in the elderly although its moisture content and cohesiveness are reduced coupled with an increase in renewal time of damaged stratum corneum... Human epidermis is highly proliferative but in a steady-state condition dependent, as are other self-renewing structures, on slowly cycling, undifferentiated stem cells. These stem cells are located within the basal compartment of the epidermis – the nonserrated keratinocytes at the tips of the epidermal rete ridges. Loss of rete ridges and consequent flattening of the dermal–epidermal junction is a hallmark of intrinsically aged skin. Such flattening results in a reduction in mean surface area of the dermal–epidermal junction. One study has estimated a reduction in mean area of dermal–epidermal junction/mm2 from 2.6 at age interval 21-40 years to 1.9 at age interval 61-80 years. These changes are accompanied by a reduction in microvilli – cytoplasmic projections from basal keratinocytes into the dermis... The rate of epidermal renewal is reduced in the skin of individuals aged 60 years or greater... Melanocytes are decreased in number in intrinsically aged epidermis, although the estimates of this decrease vary from study to study according to the methodologies used to quantitate melanocyte numbers. This said, the reduction is in the order of 8 to 20% per decade compared to young adult skin... The number of Langerhans cells is reduced in intrinsically aged epidermis... Gilchrest et al. demonstrated that subjects aged 62 to 86 years had a 42% reduction in the number of Langerhans cells in sun-protected skin as compared to young subjects aged 22 to 26 years... Numbers of dermal fibroblasts decrease with age... Aged skin is relatively hypovascular, particularly due to loss of small capillaries that run perpendicular to the dermal–epidermal junction and form capillary loops. This loss is concomitant with the loss of epidermal rete ridges. Blood vessels within the reticular dermis are reduced in number and their walls are thinned... There is an approximate 50% reduction in numbers of mast cells in intrinsically aged skin... Eccrine glands are reduced in number and function in aged skin... Age probably reduces and disorganizes the nerve supply of the skin; indeed, there is an approximate two-thirds reduction in numbers of Pacinian and Meissner’s corpuscles with age... Hair, particularly scalp hair, is lost with age in both sexes... Nails grow more slowly in the elderly... The study of aging skin particularly as a consequence of the ready accessibility of cutaneous tissue is one that presents a paradigm for aging of other organs” [44]. In derma, as a likely consequence of the exhaustion of specific stem cells, a general reduction of all its components (melanocytes, Langerhans cells, dermal fibroblasts, capillaries, blood vessels within the reticular dermis, mast cells, eccrine glands, hair, etc.) is reported and nails grow more slowly [44].
Orofacial tissues and organs. “Atrophy of the fascial planes within the eyelids may lead to herniation of the orbital fat into the lid tissue, producing the “bags under the eyes” frequently seen in the elderly. Atrophy or disinsertion of the aponeurosis of the levator palpebrae muscle, which ordinarily supports the upper eyelid, may cause the opened lid to fail to uncover the pupil, as seen in senile ptosis, despite normal levator muscle function... Secretory function of the lacrimal glands declines with age...” [45].
“Structural changes in human oral epithelia with aging include thinning the epithelial cell layers (e.g. thinning of the lingual epithelial), diminished keratinization, and simplification of epithelial structure... Histological studies of aging salivary glands show a gradual loss of acinar elements, a relative increase in the proportion of ductal elements, an increase in inflammatory infiltrates, and an increase in fibrofatty tissue” [46]. “The number of taste buds decreases after age 45, resulting in a decrease in taste sensation...” [47].
Hematopoietic cells. “...Peripheral blood lymphocyte populations do seem to show a significant change in age, with a fall in total numbers. CD4+ T-helper cells, responsible for major histocompatibility complex class II restricted recognition of foreign antigen and subsequent activation of CD8+ T-suppressor, B-lymphocyte, and granulocyte effector cells of the immune response, show an overall decline with age accompanied by a reduction in capacity to produce virgin CD4+ CD45RA T cells... Gradual involution of red marrow continues but is especially marked after the age of 70 years when iliac crest marrow cellularity is reduced to about 30% of that found in young adults” [48]. In older people, fewer neutrophils arrive at the skin abrasion sites [49]. T-lymphocytes proliferative capacity for nonspecific mitogens shows an age-related reduction [50, 51].
Age-related functional decline in hematopoietic stem cells is a likely limiting factor for longevity in mammals [52].
Gastrointestinal system. In aged individuals, various types of stomach cells show progressively shorter telomeres, and atrophy of gastric mucosa, with or without Helicobacter pylori infection, is associated with shorter telomeres [17].
“Using postmortem material, Chacko et al. (1969) found that in an Indian population the shape of villi changed on aging. The youngest subjects had finger-shaped villi, but the frequency of broad villi and convolutions increased in specimens from older people. Webster and Leeming (1975) described similar changes when fresh jejunal specimens from geriatric patients were compared with normal young controls. They found that in the elderly broader villi were more common, and in addition the villi were significantly shorter... Andrew and Andrew (1957) noticed an increase in the amount of fibrous tissue between the crypts of Lieberkuhn and a general reduction of cellularity in older mice... Lesher, Fry and Kohn (1961), Lesher and Sacher (1968) and Fry, Lesher and Kohn (1961), using autoradiography and tritiated thymidine, showed a prolonged generation time for duodenal crypt cells in old animals and an increased cell transit time (for cells to progress from the crypts to villous tips). In conclusion, the possible expected age changes in the small bowel of man are an increase in broad villi, with a reduction in villous height. These changes may be due to reduced cell production” [53].
In the colon, atrophy of the muscolaris propria and an increase in the amount of fibrosis and elastin has been shown [54].
In each intestinal crypt, there are four to six stem cells that with their intensive duplication activity renew continuously the epithelium of the small intestine [55]. The above said changes, surely due to a declining mitotic activity of crypt stem cells, as hypothesized from a long time [53], reduce intestinal functionality and, likely, overall fitness.
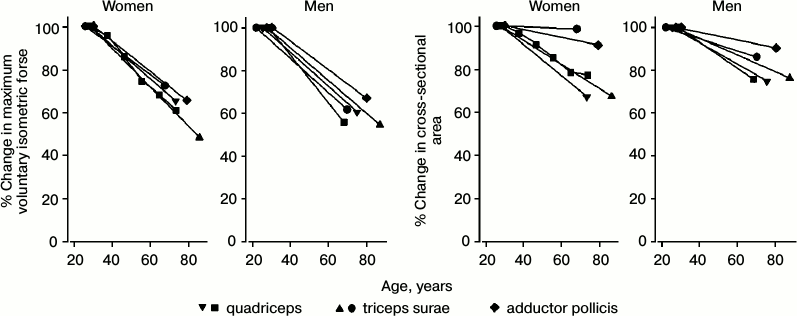
Skeletal muscle. Age-related muscle atrophy, both in terms of overall muscle bulk and of the size of individual fibers, is well known [56, 57] (Fig. 4). “These changes are to some extent dependent on the fallout of anterior horn cells that occurs with age, but this does not completely explain the process of aging atrophy. In detailed studies, it has been shown that the progressive reduction that occurs in muscle volume with aging can be detected from age 25 years and that up to 10% of muscle volume is lost by age 50 years. Thereafter the rate of muscle volume atrophy increases, so that by 80 years almost half the muscle has wasted... Reductions both in fiber number and fiber size are implicated in the loss of muscle volume” [58].
In Duchenne muscular dystrophy, chronic destruction of myocytes, continually replaced by duplication of stem cells until these are exhausted, has been described [59].
Fig. 4. Age-related decline in maximum voluntary isometric force and in cross-sectional area for various muscles [60-66].
Liver. The volume of liver shows age-related declines [67], both in proportion to body weight and in absolute values [68]. This reduction has been estimated to be about 37% between ages 24 and 91 [67]. Liver blood flow also declines with age, by about 53% between ages 24 and 91 [67] but, while liver size declines with age, hepatocytes increase in size, unlike in the liver atrophy caused by starvation [69, 70].
The chronic destruction of hepatocytes by hepatitis, alcoholism, or other factors is a known cause of cirrhosis: by exhaustion of duplication capacities of hepatocyte stem cells, the atrophic syndrome transforms the liver, often with the complication of hepatocarcinoma caused by dysfunctional telomere-induced instability [30, 71].
Pancreatic β-cells. Pancreatic β-cells show turnover [72] and an insufficient substitution of β-cells exhausted by metabolic stress has been suggested as cause of type 2 diabetes mellitus [73, 74]. Diabetes is a manifestation of Werner syndrome [75], as a likely consequence of an insufficient replacement of apoptotic β-cells by impaired replication of β-cell stem cells. Diabetes frequency shows an age-related increment [76] likely caused by the progressive exhaustion of β-cell turnover.
Drugs that are effective in “organ protection”, as ACE-inhibitors and sartans and statins, reduce the risk of diabetes [77, 78].
Bone. “Once middle age is reached, the total amount of calcium in the skeleton (i.e. bone mass) starts to decline with age... This is associated with changes in skeletal structure, resulting in it becoming weaker and more prone to sustaining fractures. For example, the bony cortex becomes thinner due to expansion of the inner medullary cavity, the trabecular network disintegrates, and there is an accumulation of microfractures... Bone loss in the elderly is largely a result of excess osteoclast activity, which causes both an expansion in the total number of remodeling sites and an increase in the amount of bone resorbed per individual site... Bone loss in the elderly is also thought to involve an age-related decline in the recruitment and synthetic capacity of osteoblasts” [79].
“Involutional bone loss... starts between the ages of 35 and 40 in both sexes, but in women there is an acceleration of bone loss in the decade after menopause. Overall, women lose 35 to 50% of trabecular and 25 to 30% of cortical bone mass with advancing age, whereas men lose 15 to 45% of trabecular and 5 to 15% of cortical bone... Bone loss starts between the ages of 35 and 40 years in both sexes, possibly related to impaired new bone formation, due to declining osteoblast function” [80].
Lungs. “The most important age-related change in the large airways is a reduction in the number of glandular epithelial cells... The area of the alveoli falls and the alveoli and alveoli ducts enlarge. Function residual capacity, residual volume, and compliance increase...” [81]. Lung volumes (FEV1, FVC) show an age-related reduction [82].
Statins contrast the decline in lung function and their anti-inflammatory and antioxidant properties could explain this effect [83]. Alternatively, it could be the consequence of actions on type II alveolar epithelial cells analogous to those on endothelial cells [31].
Kidneys. “Age-induced renal changes are manifested macroscopically by a reduction in weight of the kidney and a loss of parenchymal mass. According to Oliver, the average combined weight of the kidneys in different age groups is as follows: 60 years, 250 g; 70 years, 230 g; 80 years, 190 g. The decrease in weight of the kidneys corresponds to a general decrease in the size and weight of all organs. Microscopically, the most impressive changes are reductions in the number and size of nephrons. Loss of parenchymal mass leads to a widening of the interstitial spaces between the tubules. There is also an increase in the interstitial connective tissue with age. The total number of identifiable glomeruli falls with age, roughly in accord with the changes in renal weight” [84].
Microalbuminuria, a reliable marker of nephropathy, is “predictive, independently of traditional risk factors, of all-cause and cardiovascular mortality and CVD events within groups of patients with diabetes or hypertension, and in the general population... It may... signify systemic endothelial dysfunction that predisposes to future cardiovascular events” [85].
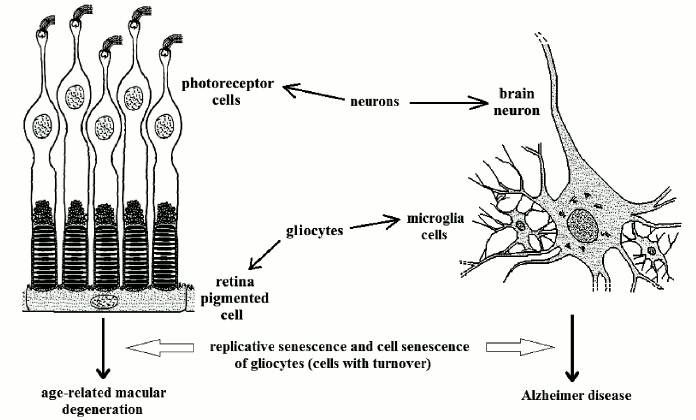
Cell types without turnover. Photoreceptor cells. Photoreceptor cells (cones and rods) are highly differentiated nervous cells without turnover, but metabolically depending on other cells with turnover, retina pigmented cells (RPCs), which are highly differentiated gliocytes.
Each day, with an extraordinary metabolic activity, every RPC phagocytizes about 10% of the membranes with photopsin molecules of about 50 photoreceptor cells. With the age-related decline of RPC turnover, the deficiency of their function kills the photoreceptors not served [86]. This is above all manifested in the functionality of the more sensitive part of the retina, the macula, from which the name “age-related retina macular degeneration” (ARMD) [87]. ARMD affects 5, 10, and 20% of subjects of 60, 70, and 80 years old, respectively [86], and it is likely that a large proportion of older individuals suffer from ARMD.
Risk factors for endothelial cells as smoking, diabetes, and obesity are risk factors for ARMD too [88].
Neurons of the central nervous system. Neurons are perennial cells, but their vitality depends on other cells (e.g. microglia, a type of gliocytes), which show turnover. The hypothesis that Alzheimer disease (AD) is caused by cell senescence of microglia cells has been proposed [10, 22, 89, 90].
Microglia cells degrade β-amyloid protein [91, 92] and this function is known to be altered in AD [93] with the consequent noxious accumulation of the protein.
Telomeres have been shown to be significantly shorter in patients with probable AD than in apparently healthy control subjects [94]. AD could have, at least partially, a vascular etiology due to age-related endothelial dysfunction [22], but “a cell senescence model might explain Alzheimer dementia without primary vascular involvement” [22].
An interesting comparison between AD and ARMD is possible: both are probably determined by the death of cells with no turnover as a likely consequence of the age-related failure of cells with turnover (Fig. 5) [10]. Moreover, AD shows an age-related increasing frequency as ARMD: it affects 1.5% of USA and Europe population at age 65 years and 30% at 80 [95] and a centenarian has a high probability of suffering from it.
Crystalline lens. The crystalline lens has no cell in its core, but its functionality depends on lens epithelial cells that show turnover [96]. “Many investigators have emphasized posttranslational alterations of long-lived crystalline proteins as the basis for senescent ocular cataracts. It is apparent in Werner syndrome that the cataracts result from alterations in the lens epithelial cells” [75], which is consistent with age-related reduction in growth potential for lens epithelial cell reported for normal human subjects [96]. Statins lower the risk of nuclear cataract, the most common type of age-related cataract [97]. This has been attributed to “putative antioxidant properties” [97], but could be the consequence of effects on lens epithelial cells analogous to those on endothelial cells [31].
Fig. 5. Some retina photoreceptors and a brain neuron (both specialized neurons) served by two types of differentiated gliocytes (RPCs and microglia cells, respectively). Cell senescence of RPCs and microglia cells cause ARMD and AD, respectively.
Other organs or tissues. Telomere dysfunction for cells in replicative senescence, in particular those, mostly epithelial, with higher turnover, is a significant cause of cancer in older individuals [30].
Finally, we must consider the numberless complications for many organs deriving by the progressive impairment of endothelial, neuronal, and immunological functions and, in general, by the interlacement of the decline of several functions [33].
PATHOLOGY OF AGING PHENOMENON
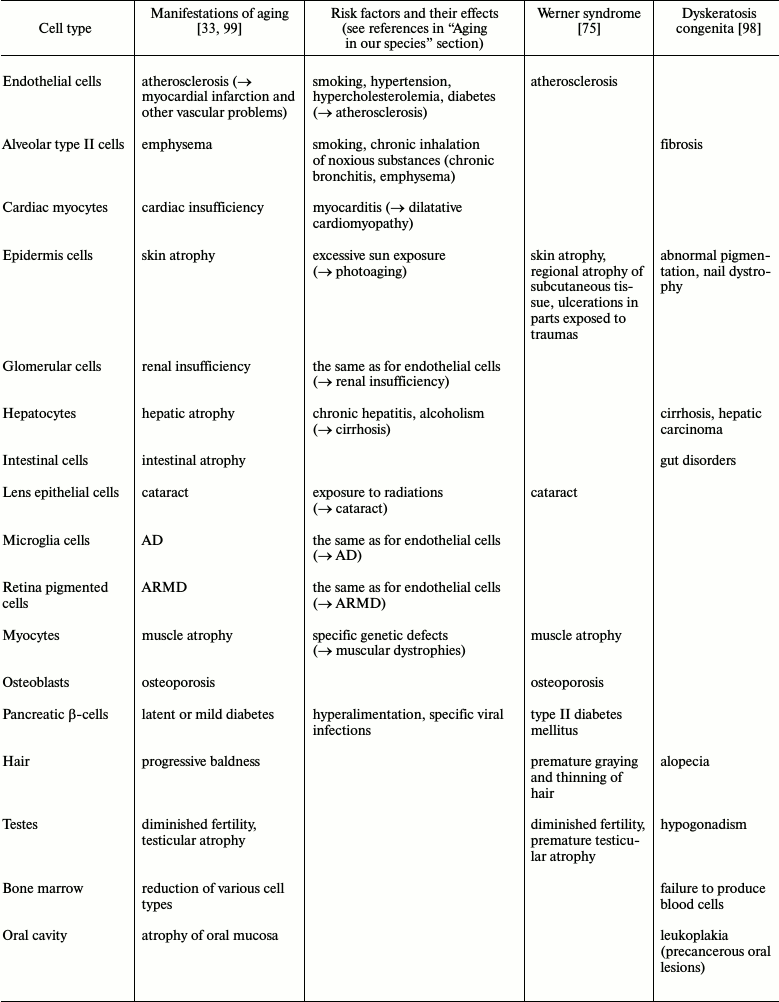
As any function, aging must have its pathology, e.g. (1) various non-genetic factors increase apoptotic rates and therefore aging manifestations (see “risk factors” in the previous section); (2) among various genetic diseases that display aging characteristics, two are particularly interesting: dyskeratosis congenita (DC) and Werner syndrome (WS). In DC, there is an inherited defect in telomerase function and “problems trend to occur in tissues in which cells multiply rapidly – skin, nails, hair, gut and bone marrow... People with DC, as well as late-generation telomerase-deficient mice, also suffer from a higher rate of cancer. This can likewise be explained by the lack of telomerase, which result in unstable chromosomes – in DC sufferers and the mutant mice, many chromosomes fuse end to end, probably because their telomeres are terminally eroded (de Lange and Jacks, 1999)” [98]. In DC, there is no alteration for tissues with no telomerase expression. On the contrary, in WS, there are problems for DNA-recombination process and so for telomere maintaining, and the disease shows alterations for tissues with lower cell turnover and a closer version of normal aging [75, 98]. These facts are summarized in the table.
CONCLUSION
Manifestations of aging and its pathologies
More than a century ago, Weismann proposed that aging was determined by an evolved limitation in the replicative capacities of cells [100]. As well discussed elsewhere [101], the famous wrong experiments of Carrel and Ebeling, the uncertainties and contradictions of the same Weismann, and various theoretical objections wiped out the scientific memory of this hypothesis.
In 1959, the great physicist Leo Szilard proposed that, by action of some factors, the somatic cells decrease with age at an accelerating rate and that this was the key factor in aging [102]. In modern times, this idea has been developed and strengthened by experimental data [103].
This shows that the importance of the relationship between aging and age-related loss of cellularity has already been pointed out by others. However, the key question is whether this correlation and its causes are in support or against, compatible or incompatible with each of the two opposing paradigms.
Programmed cell death, “on/off” and “gradual” cell senescence, cell duplication limits (variable, according to cell types and influenced by various physiological and pathological events), cell turnover and its limitations (variable, depending on cell types) are all phenomena genetically determined and regulated (with clear differences among the species).
Some features of these phenomena have no justification in terms of physiological factors other than as aging determinants. In particular, the supporters of the old paradigm try to justify the limits in cell replication as a general defense against cancer [104, 105].
But: (i) species with negligible senescence (i.e. with individuals showing no age-related decay) have no age-related reduction of telomerase activity and no increase in mortality due to cancer [8]; (ii) in the human species, studied under natural conditions, fitness decay/mortality increment (i.e. aging) reaches significant levels without a contemporaneous detectable incidence of cancer mortality. It is untenable that a defense against cancer kills large part of the population before cancer as cause of death becomes detectable [106].
Moreover, the above-said justification is even more unlikely to explain: (i) the “on/off” cell senescence program and its damaging effects; (ii) the regulatory functions of subtelomeric DNA (a condition indispensable for “gradual” cell senescence), i.e. the position of pivotal parts of DNA where they are more vulnerable when telomere shortens.
The mechanisms, genetically determined and regulated, here summarized, are a likely cause of the age-related progressive deterioration of all functions, namely aging. Their existence is predicted by the new paradigm and indeed is essential for its validity.
On the contrary, they are not expected by the old paradigm and are in complete contrast with it.
The explanation of aging through the new paradigm allows: (i) a rational and consistent interpretation of the manifestations of aging, its pathologies included (e.g. AD, ARMD, dyskeratosis congenita, Werner syndrome); (ii) the prospect of being able to change aging manifestations and even to obtain a full control of aging through scientific procedures that are technically feasible [10, 107]. The exposition and discussion of this last prospect, already briefly expounded elsewhere [107], is however outside and beyond the limits of time and of topic of this work.
REFERENCES
1.Goldsmith, T. (2013) The Evolution of Aging,
3rd Edn., Azinet Press, USA.
2.Kirkwood, T. B. L., and Austad, S. N. (2000) Why do
we age? Nature, 408, 233-238.
3.Libertini, G. (1988) An adaptive theory of the
increasing mortality with increasing chronological age in populations
in the wild, J. Theor. Biol., 132, 145-162.
4.Skulachev, V. P. (1997) Aging is a specific
biological function rather than the result of a disorder in complex
living systems: biochemical evidence in support of Weismann’s
hypothesis, Biochemistry (Moscow), 62, 1191-1195.
5.Libertini, G. (2012) Classification of phenoptotic
phenomena, Biochemistry (Moscow), 77, 707-715.
6.Finch, C. E. (1990) Longevity, Senescence and
the Genome, University of Chicago Press, London.
7.Skulachev, V. P. (2002) Programmed death phenomena:
from organelle to organism, Ann. N. Y. Acad. Sci., 959,
214-237.
8.Libertini, G. (2008) Empirical evidence for various
evolutionary hypotheses on species demonstrating increasing mortality
with increasing chronological age in the wild, Sci. World J.,
8, 183-193.
9.Kerr, J. F. R., Wyllie, A. H., and Currie, A. R.
(1972) Apoptosis: a basic biological phenomenon with wide-ranging
implications in tissue kinetics, Br. J. Cancer, 26,
239-257.
10.Libertini, G. (2009) The role of
telomere–telomerase system in age-related fitness decline, a
tameable process, in Telomeres: Function, Shortening and Lengthening
(Mancini, L., ed.) Nova Science Publishers, New York, pp.
77-132.
11.Reed, J. C. (1999) Dysregulation of apoptosis in
cancer, J. Clin. Oncol., 17, 2941-2953.
12.Alberts, B., Bray, D., Hopkin, K., Johnson, A.,
Lewis, J., Raff, M., Roberts, K., and Walter, P. (eds.) (2013)
Essential Cell Biology, 4th Edn., Garland Science, New York.
13.Anversa, P., Kajstura, J., Leri, A., and Bolli,
R. (2006) Life and death of cardiac stem cells, Circulation,
113, 1451-1463.
14.Richardson, B. R., Allan, D. S., and Le, Y.
(2014) Greater organ involution in highly proliferative tissues
associated with the early onset and acceleration of ageing in humans,
Exp. Gerontol., 55, 80-91.
15.Hayflick, L., and Moorhead, P. S. (1961) The
serial cultivation of human diploid cell strains, Exp. Cell
Res., 25, 585-621.
16.Olovnikov, A. M. (1973) A theory of marginotomy:
the incomplete copying of template margin in enzyme synthesis of
polynucleotides and biological significance of the problem, J.
Theor. Biol., 41, 181-190.
17.Takubo, K., Aida, J., Izumiyama-Shimomura, N.,
Ishikawa, N., Sawabe, M., Kurabayashi, R., Shiraishi, H., Arai, T., and
Nakamura, K-I. (2010) Changes of telomere length with aging,
Geriatr. Gerontol. Int., 10, S197-206.
18.Blackburn, E. H., and Gall, J. G. (1978) A
tandemly repeated sequence at the termini of the extrachromosomal
ribosomal RNA genes in Tetrahymena, J. Mol. Biol.,
120, 33-53.
19.Greider, C. W., and Blackburn, E. H. (1985)
Identification of a specific telomere terminal transferase activity in
Tetrahymena extracts, Cell, 51, 405-413.
20.Van Steensel, B., and de Lange, T. (1997) Control
of telomere length by the human telomeric protein TRF1, Nature,
385, 740-743.
21.Blackburn, E. H. (2000) Telomere states and cell
fates, Nature, 408, 53-56.
22.Fossel, M. B. (2004) Cells, Aging and Human
Disease, Oxford University Press, New York.
23.Ben-Porath, I., and Weinberg, R. (2005) The
signals and pathways activating cellular senescence, Int. J.
Biochem. Cell. Biol., 37, 961-976.
24.Bodnar, A. G., Ouellette, M., Frolkis, M., Holt,
S. E., Chiu, C. P., Morin, G. B., Harley, C. B., Shay, J. W.,
Lichtsteiner, S., and Wright, W. E. (1998) Extension of life-span by
introduction of telomerase into normal human cells, Science,
279, 349-352.
25.Counter, C. M., Hahn, W. C., Wei, W., Caddle, S.
D., Beijersbergen, R. L., Lansdorp, P. M., Sedivy, J. M., and Weinberg,
R. A. (1998) Dissociation among in vitro telomerase activity,
telomere maintenance, and cellular immortalization, Proc. Natl.
Acad. Sci. USA, 95, 14723-14728.
26.Vaziri, H. (1998) Extension of life span in
normal human cells by telomerase activation: a revolution in cultural
senescence, J. Anti-Aging Med., 1, 125-130.
27.Vaziri, H., and Benchimol, S. (1998)
Reconstitution of telomerase activity in normal cells leads to
elongation of telomeres and extended replicative life span, Curr.
Biol., 8, 279-282.
28.De Lange, T., and Jacks, T. (1999) For better or
worse? Telomerase inhibition and cancer, Cell, 98,
273-275.
29.Jaskelioff, M., Muller, F. L., Paik, J. H.,
Thomas, E., Jiang, S., Adams, A. C., Sahin, E., Kost-Alimova, M.,
Protopopov, A., Cadinanos, J., Horner, J. W., Maratos-Flier, E., and
Depinho, R. A. (2011) Telomerase reactivation reverses tissue
degeneration in aged telomerase-deficient mice, Nature,
469, 102-106.
30.DePinho, R. A. (2000) The age of cancer,
Nature, 408, 248-254.
31.Hill, J. M., Zalos, G., Halcox, J. P. J.,
Schenke, W. H., Waclawiw, M. A., Quyyumi, A. A., and Finkel, T. (2003)
Circulating endothelial progenitor cells, vascular function, and
cardiovascular risk, N. Engl. J. Med., 348, 593-600.
32.Werner, N., Kosiol, S., Schiegl, T., Ahlers, P.,
Walenta, K., Link, A., Bohm, M., and Nickenig, G. (2005) Circulating
endothelial progenitor cells and cardiovascular outcomes, N. Engl.
J. Med., 353, 999-1007.
33.Tallis, R. C., Fillit, H. M., and Brocklehurst,
J. C. (eds.) (1998) Brocklehurst’s Textbook of Geriatric
Medicine and Gerontology, 5th Edn., Churchill Livingstone, New
York.
34.Davidson, M. H. (2007) Overview of prevention and
treatment of atherosclerosis with lipid-altering therapy for pharmacy
directors, Am. J. Manag. Care, 13, S260-269.
35.Weir, M. R. (2007) Effects of
rennin–angiotensin system inhibition on end-organ protection: can
we do better? Clin. Ther., 29, 1803-1824.
36.Urbanek, K., Quaini, F., Tasca, G., Torella, D.,
Castaldo, C., Nadal-Ginard, B., Leri, A., Kajstura, J., Quaini, E., and
Anversa, P. (2003) Intense myocyte formation from cardiac stem cells in
human cardiac hypertrophy, Proc. Natl. Acad. Sci. USA,
100, 10440-10445.
37.Leri, A., Barlucchi, L., Limana, F., Deptala, A.,
Darzynkiewicz, Z., Hintze, T. H., Kajstura, J., Nadal-Ginard, B., and
Anversa, P. (2001) Telomerase expression and activity are coupled with
myocyte proliferation and preservation of telomeric length in the
failing heart, Proc. Natl. Acad. Sci. USA, 98,
8626-8631.
38.Chimenti, C., Kajstura, J., Torella, D., Urbanek,
K., Heleniak, H., Colussi, C., Di Meglio, F., Nadal-Ginard, B.,
Frustaci, A., Leri, A., Maseri, A., and Anversa, P. (2003) Senescence
and death of primitive cells and myocytes lead to premature cardiac
aging and heart failure, Circ. Res., 93, 604-613.
39.Olivetti, G., Melissari, M., Capasso, J. M., and
Anversa, P. (1991) Cardiomyopathy of the aging human heart. Myocyte
loss and reactive cellular hypertrophy, Circ. Res., 68,
1560-1568.
40.Aronow, W. S. (1998) Effects of aging on the
heart, in Brocklehurst’s Textbook of Geriatric Medicine and
Gerontology, 5th Edn., pp. 255-262.
41.Caird, F. I., and Dall, J. L. C. (1978) The
cardiovascular system, in Textbook of Geriatric Medicine and
Gerontology (Brocklehurst, J. C., ed.) 2nd Edn., Churchill
Livingstone, New York, pp. 125-157.
42.Jibrini, M. B., Molnar, J., and Arora, R. R.
(2008) Prevention of atrial fibrillation by way of abrogation of the
rennin–angiotensin system: a systematic review and meta-analysis,
Am. J. Ther., 15, 36-43.
43.Fauchier, L., Pierre, B., de Labriolle, A.,
Grimard, C., Zannad, N., and Babuty, D. (2008) Anti-arrhythmic effect
of statin therapy and atrial fibrillation a meta-analysis of randomized
controlled trials, J. Am. Coll. Cardiol., 51,
828-835.
44.Griffiths, C. E. M. (1998) Aging of the skin, in
Brocklehurst’s Textbook of Geriatric Medicine and
Gerontology, 5th Edn., pp. 1293-1298.
45.Brodie, S. E. (1998) Aging and disorders of the
eye, in Brocklehurst’s Textbook of Geriatric Medicine and
Gerontology, 5th Edn., pp. 659-672.
46.Devlin, H., and Ferguson, M. W. J. (1998) Aging
and the orofacial tissues, in Brocklehurst’s Textbook of
Geriatric Medicine and Gerontology, 5th Edn., pp. 789-802.
47.Reinus, J. F., and Brandt, L. J. (1998) The upper
gastrointestinal tract, in Brocklehurst’s Textbook of
Geriatric Medicine and Gerontology, 5th Edn., pp. 803-826.
48.Gilleece, M. H., and Dexter, T. M. (1998) Aging
and the blood, in Brocklehurst’s Textbook of Geriatric
Medicine and Gerontology, 5th Edn., pp. 1241-1246.
49.MacGregor, R. R., and Shalit, M. (1990)
Neutrophil function in healthy elderly subjects, J. Gerontol.,
45, M55-60.
50.Schwab, R., Hausman, P. B., Rinnooy-Kan, E., and
Weksler, M. E. (1985) Immunological studies of ageing. X. Impaired T
lymphocytes and normal monocyte response from elderly humans to the
mitogenic antibodies OKT3 and Leu4, Immunology, 55,
677-684.
51.Murasko, D. M., Nelson, B. J., Silver, R.,
Matour, D., and Kaye, D. (1986) Immunologic response in an elderly
population with a mean age of 85, Am. J. Med., 81,
612-618.
52.Geiger, H., and Van Zant, G. (2002) The aging of
lympho-hematopoietic stem cells, Nat. Immunol., 3,
329-333.
53.Webster, S. G. P. (1978) The gastrointestinal
system – c. The pancreas and the small bowel, in Textbook of
Geriatric Medicine and Gerontology (Brocklehurst, J. C., ed.) 2nd
Edn., Churchill Livingstone, New York, pp. 358-368.
54.Baime, M. J., Nelson, J. B., and Castell, D. O.
(1994) Aging of the gastrointestinal system, in Principles of
Geriatric Medicine and Gerontology (Hazzard, W. R., et al., eds.)
3rd Edn., McGraw-Hill, New York, pp. 665-681.
55.Barker, N., van Es, J. H., Kuipers, J., Kujala,
P., van den Born, M., Cozijnsen, M., Haegebarth, A., Korving, J.,
Begthel, H., Peters, P. J., and Clevers, H. (2007) Identification of
stem cells in small intestine and colon by marker gene Lgr5,
Nature, 449, 1003-1007.
56.Grimby, G., Danneskiold-Samsoe, B., Hvid, K., and
Saltin, B. (1982) Morphology and enzymatic capacity in arm and leg
muscles in 78-81 year old men and women, Acta Physiol. Scand.,
115, 125-134.
57.Lexell, J., Taylor, C. C., and Sjostrom, M.
(1988) What is the cause of the ageing atrophy? Total number, size and
proportion of different fiber types studied in whole vastus lateralis
muscle from 15- to 83-year-old men, J. Neurol. Sci., 84,
275-294.
58.Cumming, W. J. K. (1998) Aging and neuromuscular
disease, in Brocklehurst’s Textbook of Geriatric Medicine and
Gerontology, 5th Edn., pp. 1115-1130.
59.Adams, V., Gielen, S., Hambrecht, R., and
Schuler, G. (2001) Apoptosis in skeletal muscle, Front. Biosci.,
6, D1-D11.
60.Young, A., Stokes, M., and Crowe, M. (1984) Size
and strength of the quadriceps muscles of old and young women, Eur.
J. Clin. Invest., 14, 282-287.
61.Young, A., Stokes, M., and Crowe, M. (1985) The
size and strength of the quadriceps muscles of old and young men,
Clin. Physiol., 5, 145-154.
62.Vandervoort, A. A., and McComas, A. J. (1986)
Contractile changes in opposing muscles of the human ankle joint with
aging, J. Appl. Physiol., 61, 361-367.
63.Davies, C. T. M., Thomas, D. O., and White, M. J.
(1986) Mechanical properties of young and elderly human muscle, Acta
Med. Scand., 711, S219-226.
64.Klitgaard, H., Mantoni, M., Schiaffino, S.,
Ausoni, S., Gorza, L., Laurent-Winter, C., Schnohr, P., and Saltin, B.
(1990) Function, morphology and protein expression of ageing skeletal
muscle: a cross-sectional study of elderly men with different training
backgrounds, Acta Physiol. Scand., 140, 41-54.
65.Rutherford, O. M., and Jones, D. A. (1992) The
relationship of muscle and bone loss and activity levels with age in
women, Age Ageing, 21, 286-293.
66.Philips, S. K., Bruce, S. A., Newton, D., and
Woledge, R. C. (1992) The weakness of old age is not due to failure of
muscle activation, J. Gerontol. Med. Sci., 47,
M45-49.
67.Marchesini, G., Bua, V., Brunori, A., Bianchi,
G., Pisi, P., Fabbri, A., Zoli, M., and Pisi, E. (1988) Galactose
elimination capacity and liver volume in aging man, Hepatology,
8, 1079-1083.
68.Wynne, H. A., Cope, L. H., Mutch, E., Rawlins, M.
D., Woodhouse, K. W., and James, O. F. (1989) The effect of age upon
liver volume and apparent liver blood flow in healthy man,
Hepatology, 9, 297-301.
69.David, H., and Reinke, P. (1988) Liver morphology
with aging, in Aging in Liver and Gastrointestinal Tracts
(Bianchi, L., Holt, P., and James, O. F. W., eds.) MTP Press,
Lancaster (UK), pp. 143-159.
70.Watanabe, T., and Tanaka, Y. (1982) Age-related
alterations in the size of human epatocytes: study of mononuclear and
binuclear cells, Virchow Arch., 39, 9-20.
71.Artandi, S. E. (2002) Telomere shortening and
cell fates in mouse models of neoplasia, Trends Mol. Med.,
8, 44-47.
72.Finegood, D. T., Scaglia, L., and Bonner-Weir, S.
(1995) Dynamics of beta-cell mass in the growing rat pancreas.
Estimation with a simple mathematical model, Diabetes,
44, 249-256.
73.Bonner-Weir, S. (2000) Islet growth and
development in the adult, J. Mol. Endocrinol., 24,
297-302.
74.Cerasi, E., Kaiser, N., and Leibowitz, G. (2000)
Type 2 diabetes and beta cell apoptosis, Diabetes Metab.,
26, 13-16.
75.Martin, G. M., and Oshima, J. (2000) Lessons from
human progeroid syndromes, Nature, 408, 263-266.
76.Harris, M. I., Hadden, W. C., Knowler, W. C., and
Bennett, P. H. (1987) Prevalence of diabetes and impaired glucose
tolerance and plasma glucose levels in U.S. population aged 20-74 yr,
Diabetes, 36, 523-534.
77.McCall, K. L., Craddock, D., and Edwards, K.
(2006) Effect of angiotensin-converting enzyme inhibitors and
angiotensin II type 1 receptor blockers on the rate of new-onset
diabetes mellitus: a review and pooled analysis,
Pharmacotherapy, 26, 1297-1306.
78.Ostergren, J. (2007) Renin–angiotensin
system blockade in the prevention of diabetes, Diabetes Res. Clin.
Pract., 78, S13-21.
79.Dieppe, P., and Tobias, J. (1998) Bone and joint
aging, in Brocklehurst’s Textbook of Geriatric Medicine and
Gerontology, 5th Edn., pp. 1131-1136.
80.Francis, R. M. (1998) Metabolic bone disease, in
Brocklehurst’s Textbook of Geriatric Medicine and
Gerontology, 5th Edn., pp. 1137-1154.
81.Connolly, M. J. (1998) Age-related changes in the
respiratory system, in Brocklehurst’s Textbook of Geriatric
Medicine and Gerontology, 5th Edn., pp. 1073-1078.
82.Enright, P. L., Kronmal, R. A., Higgins, M.,
Schenker, M., and Haponik, E. F. (1993) Spirometry reference values for
women and men 65 to 85 years of age. Cardiovascular health study,
Am. Rev. Respir. Dis., 147, 125-133.
83.Alexeeff, S. E., Litonjua, A. A., Sparrow, D.,
Vokonas, P. S., and Schwartz, J. (2007) Statin use reduces decline in
lung function, Am. J. Respir. Crit. Care Med., 176,
742-747.
84.Jassal, V., Fillit, H., and Oreopoulos, D. G.
(1998) Aging of the urinary tract, in Brocklehurst’s Textbook
of Geriatric Medicine and Gerontology, 5th Edn., pp. 919-924.
85.Weir, M. R. (2007) Microalbuminuria and
cardiovascular disease, Clin. J. Am. Soc. Nephrol., 2,
581-590.
86.Berger, J. W., Fine, S. L., and Maguire, M. G.
(1999) Age-Related Macular Degeneration, Mosby, St. Louis,
USA.
87.Fine, S. L., Berger, J. W., Maguire, M. G., and
Ho, A. C. (2000) Age-related macular degeneration, N. Engl. J.
Med., 342, 483-492.
88.Klein, R., Deng, Y., Klein, B. E., Hyman, L.,
Seddon, J., Frank, R. N., Wallace, R. B., Hendrix, S. L., Kuppermann,
B. D., Langer, R. D., Kuller, L., Brunner, R., Johnson, K. C., Thomas,
A. M., and Haan, M. (2007) Cardiovascular disease, its risk factors and
treatment, and age-related macular degeneration: women’s health
initiative sight exam ancillary study, Am. J. Ophthalmol.,
143, 473-483.
89.Fossel, M. B. (1996) Reversing Human
Aging, William Morrow and Company, New York.
90.Flanary, B. (2009) Telomeres: function,
shortening, and lengthening, in Telomeres: Function, Shortening and
Lengthening (Mancini, L., ed.) Nova Science Publishers, New York,
pp. 431-438.
91.Qiu, W. Q., Walsh, D. M., Ye, Z., Vekrellis, K.,
Zhang, J., Podlisny, M. B., Rosner, M. R., Safavi, A., Hersh, L. B.,
and Selkoe, D. J. (1998) Insulin-degrading enzyme regulates
extracellular levels of amyloid beta-protein by degradation, J.
Biol. Chem., 273, 32730-32738.
92.Vekrellis, K., Ye, Z., Qiu, W. Q., Walsh, D.,
Hartley, D., Chesneau, V., Rosner, M. R., and Selkoe, D. J. (2000)
Neurons regulate extracellular levels of amyloid beta-protein via
proteolysis by insulin-degrading enzyme, J. Neurosci.,
20, 1657-1665.
93.Bertram, L., Blacker, D., Mullin, K., Keeney, D.,
Jones, J., Basu, S., Yhu, S., McInnis, M. G., Go, R. C., Vekrellis, K.,
Selkoe, D. J., Saunders, A. J., and Tanzi, R. E. (2000) Evidence for
genetic linkage of Alzheimer’s disease to chromosome 10q,
Science, 290, 2302-2303.
94.Von Zglinicki, T., Serra, V., Lorenz, M.,
Saretzki, G., Lenzen-Grossimlighaus, R., Gessner, R., Risch, A., and
Steinhagen-Thiessen, E. (2000) Short telomeres in patients with
vascular dementia: an indicator of low antioxidative capacity and a
possible risk factor? Lab. Invest., 80, 1739-1747.
95.Gorelick, P. B. (2004) Risk factors for vascular
dementia and Alzheimer disease, Stroke, 35,
2620-2622.
96.Tassin, J., Malaise, E., and Courtois, Y. (1979)
Human lens cells have an in vitro proliferative capacity
inversely proportional to the donor age, Exp. Cell Res.,
123, 388-392.
97.Klein, B. E., Klein, R., Lee, K. E., and Grady,
L. M. (2006) Statin use and incident nuclear cataract, JAMA,
295, 2752-2758.
98.Marciniak, R., and Guarente, L. (2001) Human
genetics. Testing telomerases, Nature, 413, 370-372.
99.Fillit, H. M., Rockwood, K., and Woodhouse, K.
(eds.) (2010) Brocklehurst’s Textbook of Geriatric Medicine
and Gerontology, 7th Edn., Saunders Elsevier, Philadelphia.
100.Weismann, A. (1892) Essays upon Heredity and
Kindred Biological Problems, Vol. II, Clarendon Press, Oxford.
101.Kirkwood, T. B. L., and Cremer, T. (1982)
Cytogerontology since 1881: a reappraisal of August Weissmann and a
review of modern progress, Hum. Genet., 60, 101-121.
102.Szilard, L. (1959) On the nature of the aging
process, Proc. Natl. Acad. Sci. USA, 45, 30-45.
103.Skulachev, V. P. (2012) What is
“phenoptosis” and how to fight it? Biochemistry
(Moscow), 77, 827-846.
104.Campisi, J. (1997) The biology of replicative
senescence, Eur. J. Cancer, 33, 703-709.
105.Wright, W. E., and Shay, J. W. (2005) Telomere
biology in aging and cancer, J. Am. Geriatr. Soc., 53,
S292-294.
106.Libertini, G. (2013) Evidence for aging
theories from the study of a Hunter-Gatherer people (Ache of Paraguay),
Biochemistry (Moscow), 78, 1023-1032.
107.Libertini, G. (2009) Prospects of a longer life
span beyond the beneficial effects of a healthy lifestyle, in
Handbook on Longevity: Genetics, Diet & Disease (Bentely, J.
V., and Keller, M. A., eds.) Nova Science Publishers, New York, pp.
35-95.