REVIEW: Microbiota and Mitobiota. Putting an Equal Sign between Mitochondria and Bacteria
D. B. Zorov1*, E. Y. Plotnikov1, D. N. Silachev1, L. D. Zorova2, I. B. Pevzner3, S. D. Zorov3, V. A. Babenko3, S. S. Jankauskas3, V. A. Popkov3, and P. S. Savina4
1Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia; fax: (495) 939-0338; E-mail: zorov@genebee.msu.su; plotnikov@genebee.msu.ru2Lomonosov Moscow State University, International Laser Center, 119991 Moscow, Russia
3Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia
4Eltsyn Ural Federal University, Biological Faculty, 620002 Ekaterinburg, Russia
* To whom correspondence should be addressed.
Received June 11, 2014
The recent revival of old theories and setting them on modern scientific rails to a large extent are also relevant to mitochondrial science. Given the widespread belief that mitochondria are symbionts of ancient bacterial origin, the processes inherent to mitochondrial physiology can be revised based on their comparative analysis with possible involvement of bacteria. Such comparison combined with discussion of the role of microbiota in pathogenesis allows discussion of the role of “mitobiota” (we introduce this term) as the combination of different phenotypic manifestations of mitochondria in the organism reflecting pathological changes in the mitochondrial genome. When putting an equal sign between mitochondria and bacteria, we find similarity between the mitochondrial and bacterial theories of cancer. The presence of the term “bacterial infection” suggests “mitochondrial infection”, and mitochondrial (oxidative) theory of aging can in some way be transformed into a “bacterial theory of aging”. The possible existence of such processes and the data confirming their presence are discussed in this review. If such a comparison has the right to exist, the homeostasis of “mitobiota” is of not lesser physiological importance than homeostasis of microbiota, which has been so intensively discussed recently.
KEY WORDS: mitochondria, ultrastructure, bacteria, microbiota, mitobiota, mitohormesis, diseases, inflammation, cancer, infection, aging, death, phenoptosisDOI: 10.1134/S0006297914100046
MITOCHONDRIA. EVOLUTIONARY AND POST-EVOLUTIONARY HISTORY
The current information revolution in natural sciences allows quick finding and analyzing of old, often forgotten, theories and materials that were sometimes ignored because of insufficient development of experimental bases in the past. This results in revival and revision of these data given the current development of science. In this regard, we now have the opportunity to compare the structure and functions of mitochondria based on the preferred theory of their bacterial origin.
In the previous two issues of “Phenoptosis” (as part of the journal Biochemistry (Moscow)), we discussed the possibility that mitochondria might be responsible for the death of not only a cell, but also of an organ and the entire organism [1]. In this regard, we proposed changing the interpretation of the concept of mitochondrial medicine [2] given the significant impact of mitochondrial functioning on pathogenesis. In both articles we drew parallels between mitochondria and bacteria; we even suggested that the relationship between mitochondria and the host cell can be considered not only as symbiotic, but also as having some parasitic features (that are characteristic of bacteria) on the mitochondrial side [2].
Moreover, given the critical role of mitochondria in the organization of cell death (and maybe also the death of organs and the organism), which is not always accompanied by plans for replacement of a killed cell by a new one, such symbiosis between a mitochondrion and other cell parts (or other cells) seems at least strange.
The structures that are now called “mitochondria” were first mentioned in 1841, when Henle described some granules surrounding myofibrils in myocytes [3]. Sixteen years later the Swedish anatomist Kolliker provided a detailed description of the organization of these granules in the rows between the myofibrils in the muscle cell, calling them “blasse Kornchen”, i.e. “plane granules”. He also suggested that these granules could be involved in the metabolism of muscle fibers [4]. In 1890 Retzius called these granules “sarcosomes” [5], making a big step in distinguishing between these granules and fat droplets. In addition, similar to Cajal in 1888 [6], he observed these granules in the area of myofibrillar I disks. Later Regaud and Favre attributed these granules to mitochondria [7], after the Benda’s introduction in 1900 of the term “mitochondria” by combining the Greek words “µιτοσ” (thread) and “χονδροσ” (grain) [8]. The author of this term, which is still in use today, implemented a specific technique of mitochondrial staining that is constantly being improved at present.
The bacterial origin of mitochondria, including the concept of endosymbiosis, is now generally recognized. The endosymbiotic theory of the origin of mitochondria was put forth at the end of the XIXth and beginning of the XXth centuries, and it is rather related to chloroplasts, which, as the Russian botanist K. Merezhkovsky suggested, are also of bacterial origin [9, 10]. In principle, similar ideas were also expressed earlier, e.g. in 1883 the assumption of symbiotic relationship between chloroplasts and a cell was rather vaguely postulated by Schimper [11]. In the 1920s, Wallin called the interaction of mitochondria with a cell – symbiosis [12, 13]. This view was largely neglected and was resurrected by works of Sagan [14] and Margulis [15]. For Sagan, mitochondria were a particular example of the general theory explaining the apparent gap between prokaryotes and eukaryotes. According to this theory, the first stage of prokaryotes evolving into eukaryotes was adaptation of prokaryotes to the new oxygen-containing atmosphere due to introduction of an aerobic prokaryotic microbe (protomitochondrion) in the cytoplasm of a heterotrophic anaerobe. This resulted in the development of obligatory endosymbiosis and led to the emergence of the first aerobic amitotic amoeboid organisms around 600 million years ago. In parallel to this hypothesis, there appeared the concept of mitochondria (with their own DNA (mDNA)) as carriers of “cytoplasmic” heredity, in particular, based on the analysis of non-Mendelian distribution of mitochondria and plastids from generation to generation. Significant similarity between the chemical composition of bacteria and mitochondria and similarity of the elements of their bioenergetics are the main argument in favor of the bacterial origin of mitochondria, although a great diversity of bacterial habitats leads to similar diversity of their bioenergetics.
MITOCHONDRIAL DIVERSITY. INTRODUCTION OF THE TERM
“MITOBIOTA”
The bioenergetic diversity of mitochondria is as significant as that of bacteria. This is especially noticeable in yeast cells, which can be explained by the variety of habitats used by these eukaryotes. In the more highly organized eukaryotes, bioenergetics is not as diverse, although it is not so easy to evaluate the extent of this diversity. The diversity of mitochondrial bioenergetics in animals is not as high, although the almost anaerobic environment (in particular, in case of parasitic helminths, e.g. the roundworm) causes a situation when only part of the respiratory chain is used effectively [16].
The diversity of mitochondrial functions is clearly determined by the specialization of cells and tissues [17]. It is clear that in cells with high metabolic activity, the bioenergetic component of mitochondrial functioning acquires great significance, while in other cells alternative mitochondrial functions may prevail over bioenergetic ones (e.g. steroid synthesis in mitochondria from steroidogenic tissues or heat production in brown fat cells due to mitochondrial uncoupling protein).
It seems that genetic diversity of mitochondria by definition cannot be significant, since it is genetic conservatism of mDNA that serves as its signature of an individual and the basis for its identification. However, this is true only for young and healthy individuals. Any inherited or acquired change of mDNA (point mutations, deletions, and insertions noticeable in both homo- and heteroplasmic versions) is the basis of a number of pathologies studied in mitochondrial medicine [2].
Changes in the genetic component of mitochondria lead to obvious changes in mitochondrial ultrastructure, resulting in enormous phenotypic variability of mitochondria. Let us remember that only 13 polypeptides out of 1500 mitochondrial proteins are encoded in the mitochondrial genome, while all the other proteins are encoded in the nucleus. Hence, mitochondrial pathologies result from disorders in two genetic components of these proteins, including the processes of transcription and translation as well as posttranslational changes [2]. Figure 1 shows some examples of such phenotypic variability.
Fig. 1. Variability of normal and pathological ultrastructure of mitochondria (phenotypic variability). a) Normal ultrastructure of mitochondria in striated muscle [116] (reprinted with permission); b) intramitochondrial filaments in renal cells after administration of glycerol [117] (reprinted with permission); c) paracrystalline structures in mitochondria of endothelial cells with mitochondrial cytopathy [118] (reprinted with permission); d) prismatic cristae in oyster myocardium [119] (reprinted with permission); e) circular cristae in mitochondrion of a patient with mitochondrial myopathy and lactic acidosis [120] (reprinted with permission); f) mitochondria with circular cristae in endobronchial plasma cell granuloma [121]; g) prismatic cristae in mitochondria in a hamster brain astrocyte [122] (reprinted with permission).
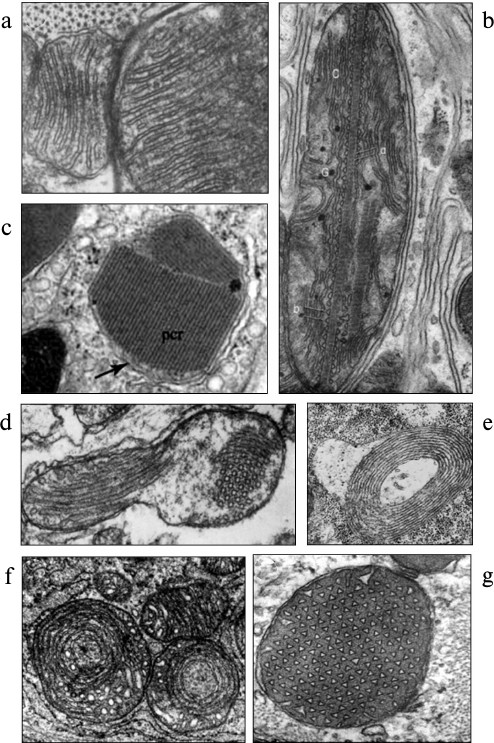
Again we admit that phenotypic variability can be determined by pathological changes resulting in the presence of different mitochondrial populations in the cell of the carrier of the pathology. Mitochondria from these populations can greatly differ in their ultrastructure. In this respect, we need to address again to the data on phenotypic expression of a number of mitochondrial diseases, when the disease becomes noticeable only after reaching a threshold of mDNA heteroplasmy (i.e. the ratio between pathologic and wild-type mDNA reaches a certain level). This implies a rather trivial conclusion that at the phenotypic (ultrastructural) level a pathology can be diagnosed based on the ratio of mitochondria with normal to changed ultrastructure. An example of such ultrastructural variability in a mitochondrial population within one cell is presented in Fig. 2 (a and b) where we can clearly see normal and changed mitochondria in the axon of a carrier of neurological pathology.
Fig. 2. Examples of pathological “mitobiota”. a) Ultrastructure of the peroneal nerve in a patient with a large mDNA deletion. The long arrow indicates the loss of cristae within the amyelinated axon. Scale, 200 nm. Small arrows show neurotubules; b) mitochondrial population within the myelinated axon of a patient with necrotizing vasculitis. Some of the mitochondria in the population have all the features of ultrastructural pathology – the matrix is lightened and cristae are absent. Scale, 1 μm ([123], reprinted with permission); c) only a fraction of cardiomyocyte mitochondria in a patient with hereditary mitochondrial hypertrophic cardiomyopathy has active cytochrome oxidase ([124], reprinted with permission).
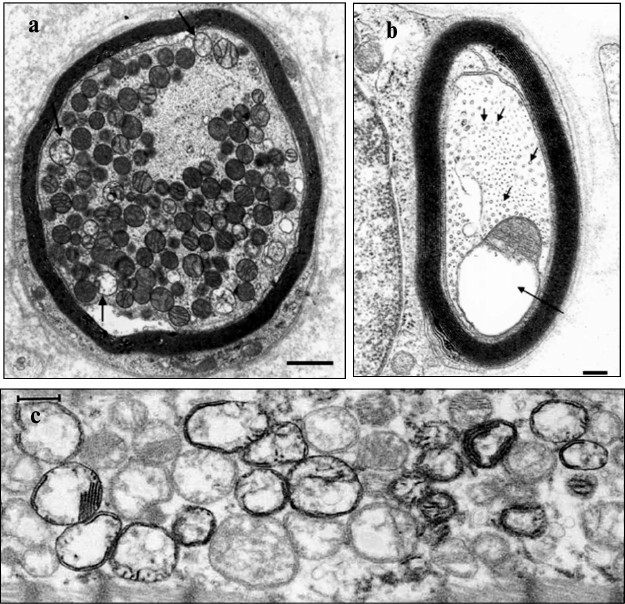
This suggests that a change in ultrastructure results from biochemical changes in mitochondria, which in turn may result from genetic changes, in particular, in the mitochondrial genome. An example of such biochemical variability in the mitochondrial population of a single cell is shown in Fig. 2c, when some mitochondria have, and others do not have active cytochrome oxidase. In this case, it results from a pathology of genetic origin. Note that there is no visually obvious ultrastructural difference between the “healthy” and “sick” mitochondria in the last example, which indicates either incomplete relations between ultrastructural and biochemical changes in mitochondria, or the imperfect indexing of mitochondrial ultrastructure. Unfortunately, we must admit the latter, because little progress has been observed in the assessment of the fine structural organization of mitochondria despite the advent of sophisticated and expensive methods. We can compare the data on ultrastructural organization of mitochondria obtained by the classical method of transmission electron microscopy more than half a century ago, when microscopic connections between cristae and the inner membrane (pedicula cristae) were first described, and similar pictures obtained now by high-resolution electron microscopy tomography (Fig. 3).
Fig. 3. Mitochondrial ultrastructure. A) Three-dimensional reconstruction of a mitochondrion from a terminal axon ([125], reprinted with permission); B) internal structure of a rat liver mitochondrion obtained by electron tomography. OM, outer membrane; IM, inner membrane; C, cristae (bold arrows indicate the contact sites between cristae and inner membrane); the triangle indicates a tubular connection between crista and the membrane ([126], reprinted with permission); C) three-dimensional reconstruction of a segment of a mouse liver parenchymal mitochondrion demonstrating the diversity of cristae form and architecture in two perpendicular planes (a – cristae mitochondreales; b – pediculus cristae; c – cristae with a set of pediculus cristae) ([127], reprinted with permission).
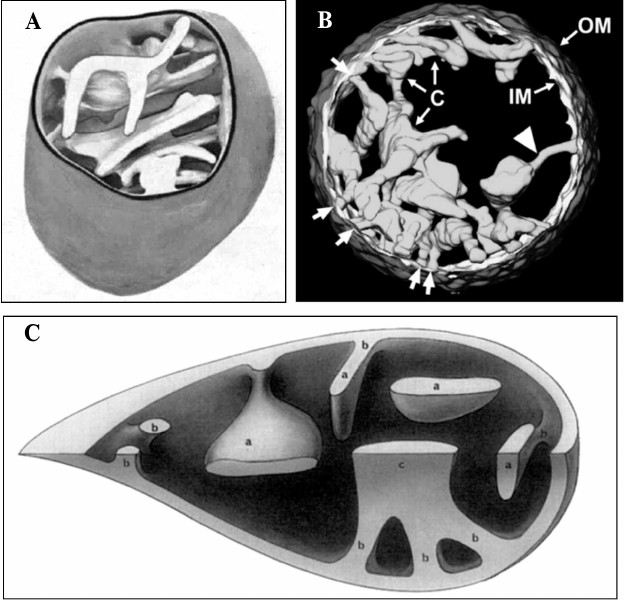
By analogy with the term microbiota representing a population of different bacteria coexisting in a certain volume, we can introduce the term mitobiota as a population of mitochondria of different structure, genetics and biochemistry coexisting within one cell (effect of “alien” mitochondria will be discussed in the section “Known Bacterial and Possible Mitochondrial Infection”). Nevertheless, “unfriendly” relationships between mitochondria are also possible; in this case they result from special programs promoting elimination of alien mitochondria, e.g. in a fertilized egg of most mammals, where only maternal mitochondria are kept, while those originating from the father are destroyed [18]. Obviously, elimination of mitochondria is one of the most important processes for the cell because long-term preservation in mitobiota of a population of mitochondria with impaired structure and functions can lead to pathological consequences for the organism. The cell constantly fights them either by controlling the quality of mitochondria and eliminating those organelles that did not pass this control [19], or by self-eliminating together with all the mitochondria following the mechanism of programmed cell death. The latter process clearly indicates the critical state of the system where the state of mitobiota transcends the allowable heterogeneity. It also points to the great importance of maintaining homeostasis of mitobiota.
BRIEFLY ON MICROBIOTA
The earliest known assumptions on the importance of microbiota were made by Hippocrates, who said “…death sits in the gut…” in 400 BC [20]. Much later, at the end of XIXth and beginning of XXth centuries Hippocrates’ thought was picked up by L. Kuhne and the Nobel Prize winner (1908) I. Metchnikoff. Kuhne believed that excessive consumption of food and its wrong composition leads to the formation of intestinal toxins [21]. Metchnikoff suggested that the digestion of milk was accompanied by introduction of desired microflora in the intestine, and that many diseases (including aging) are caused by the waste products of putrefying bacteria inhabiting the intestine. He recommended lactobacteria to fight these putrefying bacteria to eliminate this “autointoxication”. In fact, he introduced the term “dysbacteriosis” (dysbiosis) as toxic damage of the intestine (“…I attribute to the microbes of our large intestine the ability to cause premature aging and death…”) [22] due to the wrong microfloral composition and changes in the intestinal distribution of bacteria and in their metabolic activity.
“There is a widespread idea that the microbes of our intestines are in symbiotic relationship with our organism, but I think the opposite to be true”, – Mechnikov wrote in the preface to the fifth edition of his book “On the Nature of Man”. “I think that we feed a large amount of harmful microbes, which shorten our life and cause premature and painful aging”.
Now understanding of the important role of the intestine in the functioning of the organism is determined not only by its ability to carry out the degradation of food with subsequent absorption of nutrients into the blood, but also by its role as the location of the distinct immune system cohabiting with microbiota. There is also a very convincing hypothesis that bacterial pathogens are responsible for sepsis of intestinal origin and ultimately for the syndrome of systemic inflammatory response, which in most cases causes death. Violations of the permeability of intestinal epithelium lead to damaging of distant organs and, probably, this is how the syndrome of multiple organ failure develops [23]. Evidence of direct involvement of microbiota in the development of diabetes of types I and II [24-26], atherosclerosis [27], systemic inflammatory response [28], burn trauma [29-31], autism [32, 33], and allergy [34] has been proved. A remarkable review by Sekirov et al. [35] describes the network of interactions between different representatives of human microbiota and the host’s metabolism; it is noted that each member of the bacterial population occupies its own special niche in the general biochemical metabolism of a human. Hence, the importance of the balance of such interaction becomes clear, as well as the dismal result of the imbalance caused by elimination of some microbiota members from the general scheme, not to mention the replacement of representatives of one type of microbiota by others and its massive elimination due to irresponsible use of antibiotics [35].
Scientists have already started to recognize the importance of microbiota in the solution of problems with different pathologies, including aging and programmed death of an organism (phenoptosis). It should be clearly understood that the condition of the intestine is crucial when considering the problem of increasing life expectancy; it requires constant monitoring and appropriate care, which can be reduced to caring for the homeostasis of the microbiota, though it is not yet clear exactly how it can be done [23].
MITOCHONDRIAL ROLE IN INFLAMMATION
Mitochondria are the main ATP producers in cells. Cells use ATP as an energy-rich compound essential for the maintenance of a number of processes (creating gradients, mechanical movement of components of biological machines, light emission, attachment of phosphate groups to micro- and macromolecules, etc.). However, all these processes generally take place inside the cell. The intracellular content of ATP reaches millimolar concentrations, which is rather paradoxical given the micromolar affinity of ATP-consuming intracellular systems. However, earlier we proposed an explanation for this paradox by building a buffer system to maintain homeostasis of mitochondrial membrane potential [2].
The situation changes dramatically when ATP is outside the cell, where it can play the role of a modulator of the immune system, which, as we have repeatedly pointed out, plays a crucial role in a large number of pathologies, especially in sepsis, which is believed to be a killer number one. We recall that today scientists consider two types of septic response – one accompanied by bacteremia, and another one having all the signs of sepsis but with sterile blood [1]. It should be noted that the task of elimination of proinflammatory signals in the treatment of sepsis so far has been unsuccessful, with the complete failure of a number of promising drugs in the final phases of clinical trials [36]. The demonstration that elimination of extracellular ATP by apyrase prevents not only accumulation of IL-1β and production of inflammasome-independent cytokines like TNF and IL-10, but also prevents cell disintegration, mitochondrial damage, apoptosis, damage to the epithelial barrier, and even death [37], is highly promising in treatment of this critically important pathology that presumably has phenoptotic mission [38].
MITOHORMESIS
It is obvious that ATP in principle cannot be regarded as a toxic substance, because the drug, adenosine triphosphate, is recommended for clinical use in muscular dystrophy and atony, myoatrophy, multiple sclerosis, poliomyelitis, peripheral vascular diseases (Raynaud’s disease, thromboangiitis obliterans, intermittent claudication), paroxysmal supraventricular tachycardia, pigmentary retinal degeneration, paroxysm of supraventricular tachycardia, coronary heart disease, etc. [39]. A clinically recommended drug, by definition, cannot be toxic, although perhaps this requirement is in reality impracticable, because for all medications there is a toxic dose and a therapeutic permissive window of doses. It is clear that even when using small doses of a toxic compound, one can achieve not only a direct therapeutic effect, but also prime a system (adapt it) to better meet a massive attack by higher doses. There is a classic example of ischemic preconditioning, which can be reduced to a succession of short-term alternating pulses of hypoxia–reoxygenation for further less damaging response to a long hypoxic attack [40]. Similar adaptive processes occur during adaptation to low temperatures by short exposure to low temperature resulting in the development of resistance to hypothermia applied on a longer time scale [41]. Mithridates, the head of the kingdom of Pontius, is known to have constantly taken poison in small doses; and as a result, he was unable to poison himself when he wanted to. This concept of habituation to poisons is called “hormesis” in toxicology as any two-phase (in relation to dose) adaptive response (small dose heals – high dose kills). Not so long ago this concept was first applied to mitochondria under the name of “mitohormesis” [42]; it was found that mild mitochondrial stress provides resistance to diseases and death (by the way, perhaps we should reflect on the main principles of “mitohormesis” to understand the therapeutic effect of the “mild” uncoupling of oxidative phosphorylation [43, 44]). Mitochondrial oxidants (reactive oxygen species, ROS) [42] are now considered to be one of the “toxic” components of mitohormesis, but no one has yet considered DAMPs (damage associated molecular patterns) as elements of mitohormesis, i.e. those mitochondrial components that cause immune, often fatal systemic response (for explanation, see [1]).
When considering ATP as an extracellular signaling factor that can be conditionally toxic (i.e. induce processes, in particular, in the development of hyperactive immune response that will be self-damaging, resulting in the death of the organism), we should evaluate primary signaling. This is probably not too difficult on the fundamental level given the diversity of purine receptors and their abundance (purinergic receptors are the most widespread in living organisms [45]). Also, one should remember that ATP belongs to DAMPs along with other components (in particular, mitochondrial components) and is a potent modulator of various types of cell activity [46]. The complete signaling function of extracellular ATP remains to be revealed.
To re-create a full picture of possible bacterial incidence of any pathology, we should not forget that bacterial proteins, due to rather high homology with mitochondrial proteins, could quite easily be transported into mitochondria [47, 48]. Thus, one can speculate about some unfriendly signaling “talk” in protein language between the present and former bacteria (mitochondria). Such signaling leads to the situation when the former bacteria, having received such a signal, eventually trigger the program of cell death [49]. This can be regarded as a kind of antagonism between the former and present bacteria, leading to the suicide of the cell together with bacteria that have penetrated it, to prevent the spread of infection (as a version of the samurai law “it is better to die that to make a mistake” [50]).
BACTERIAL ORIGIN OF CANCER
Already in the XVIIIth century, scientists were thinking on the bacterial nature of cancer: lung cancer was believed to develop in places with scars formed in the lungs after tuberculosis [51]. Mycobacterium tuberculosis was suggested to be the cause of carcinoma (the history of this connection between cancer and tuberculosis is summarized in [52] and presented in detail in [53]). Almost a century later, the founder of modern pathological anatomy R. Virchow noticed the connection between the high frequency of bladder cancer in patients infected with Schistosoma haemotobium [54, 55]. He also discovered the presence of lymphocytes in cancerous tissues [55], which has become the basis of a theory of cancer. When analyzing the above two publications, we conclude that Virchow was the first to show the direct logical connection between carcinogenesis and inflammation initiated by bacterial toxins [56-58].
Perhaps, W. Russell, working as a pathologist at the medical school in Edinburgh, was the first to suggest the presence of parasites in cancer cells (he also thought that he had found them). Russell presented a report at a meeting of the Society of Pathologists in London on December 3, 1890. He had discovered rounded parasites living inside and outside the cancer cell; their size was about half the size of an erythrocyte. Russell called them blastomycetes, considering them as the source of the disease. His contemporaries believed that Russell observed the products of cell degradation, but the name “Russell bodies” has stuck. However, in 1901 Harvey Gaylord, working at the University of Buffalo, New York state, confirmed the data of Russell [59], having found in each of the examined cancer cells Russell bodies sized from that of an ordinary staphylococcus up to structures of ~50-µm size. However, in the early XXth century most oncologists rejected “cancer parasites” as the cause of cancer, although the parasitic theory of cancer was periodically reconsidered and this story can be found in the book “The Cancer Microbe” by Alan Cantwell published in 1990. Other stories about similar type of studies and approaches can be found in the book “Can Bacteria Cause Cancer?” by D. Hess published in 1997. Of the persons described in these books, we would like to mention Wilhelm Reich, who believed the bacteria that he called “T-bacilli” (in the 1930s) were present not only in cancer cells, but also in the blood of cancer patients, and four women – Virginia Livingston (who discovered the method of staining of what she believed to be cancer microbes), then Eleanor Alexander Jackson, Irene Diller, and Florence Siebert – they all believed the cancer microbe to be pleomorphic (i.e. having diverse phenotype) and filterable, i.e. thought it to be similar to a viral particle at a certain stage of its life cycle. It was believed that these microbes (known as mycoplasma) had no cell wall and could take the form of cocci, but had the ability to significantly increase in size. Yet we should admit that most of the opinions of these scientists and doctors were naive, not based on accurate data, and were not taken seriously by the scientific community. For example, the work of Milton Wainwright published in 2006 in Current Trends in Microbiology [60], which suggested the potential carcinogenic role of nanomicroorganisms not of viral nature, is not even mentioned in PubMed.
However, this bacterial concept of the origin of cancer was not completely dismissed, and the history of cancer microbiology has already become quite rich. The international medical community does not accept this perspective, and Stone Friedberg, who had discovered in the 1940s the S-shaped bacterium in the stomach of an ulcer patient (this bacterium is now known as Helicobacter pylori), was that time alone. Nobody believed him because doctors dogmatically thought that bacteria could not live in such high acidity. Friedberg’s supervisor demanded that he quit the project and he gave up his research. Nevertheless, in 2005 two Australian scientists (Barry Marshall and Robin Warren) were awarded a Nobel Prize for proving the bacterial nature of ulcer. And it was that very H. pylori that was shown to cause ulcer. Over the last 10 years, there has been accumulated strong evidence that these bacteria can be considered as the main cause of not only ulcer, but also stomach cancer [61, 62] and mucosal layer-mediated β-cell lymphoma [63]. Currently, the connection between these cancer types and bacterial infection is proven. In addition, at this point it is probably the only proven connection, while for other infections the causal relationship is not so obvious, and we can talk only about the association of a particular infection with the development of a corresponding cancerous disease.
Another pathogen, Streptococcus bovis, is associated with the development of cancer. It is often found in human intestinal microflora. However, the connection between this pathogen and cancer is not direct – it is mediated by infection-induced endocarditis. Most patients with endocarditis are diagnosed also with colon cancer, probably resulting from chronic inflammation [64]. There is evidence that Chlamydia pneumoniae infection dramatically increases the risk of lung cancer [65, 66]. There are also other observations of the connection between infection and the development of cancer that are systematized in [67]. In short, the bacterial theory of cancer exists, and it is rather solidly grounded [68]. There is also another idea, the viral theory of cancer but it will not be described here because it does not fit into the context and ideology of our discussion of the bacterial–mitochondrial theory of cancer.
MITOCHONDRIAL THEORY OF THE ORIGIN OF CANCER
A large number of nonspecific causes, such as radiation, chemical agents, viruses, inflammation, etc. are the basis of very specific processes that determine malignant transformation. The eminent biochemist Albert Szent-Gyorgyi wrote: “It is getting more and more difficult to find something that is not carcinogenic”; Szent-Gyorgyi described this phenomenon as the “oncogenic paradox” [69], which remained unresolved. Understanding the ambiguity of apparent multiplicity of the causes of cancer finally reaching the identical result, there is constantly search for a single principle, or rather the primary target, which in the end determines the development of cancer.
It is clear that cells need to produce enough energy to maintain vitality and perform functions determined by the genome. This obvious fact repeatedly forced researchers to look at mitochondria as the source of energy required for cell transformation followed by hardly controlled proliferation.
In today’s literature, Otto Warburg is believed to be the first to suggest that mitochondria are involved in carcinogenesis. He discovered that cancer could be caused by a lack of oxidative phosphorylation [70] (Nobel Prize in 1931). Cancer cells produce energy via glycolysis even at high oxygen concentrations. That is why they are characterized by glycolytic phenotype. This could probably be explained by the fact that other sources and suppliers of energy are unreliable in cancer cells. Hence, according to Warburg’s followers, one of the reasons for neoplastic transformation should be searched for in mitochondria.
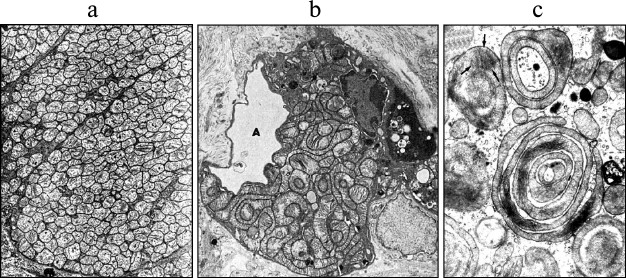
Indeed, defects in mitochondria cause a number of genetic and acquired pathologies [71]. Mitochondria are critical elements of bioenergetics, anabolism, the cell death [72], and violation of any of these processes can cause serious disorders of the vital functions of cells. In the case of oncogenesis, this means that some damage of mitochondrial structure and functions is a prerequisite of malignant transformation, and this hypothesis has been confirmed [73, 74]. Now there is clear understanding that mitochondria of cancer cells are structurally and functionally abnormal, at least in terms of energy production. Such mitochondria have a phenotype different from mitochondria of normal cells, in particular with regard to the set of proteins and lipids (summarized in [75]). It has been long observed that the development of malignancy and aerobic glycolysis is preceded by the loss of mitochondrial functions [76-79], and it determines genomic instability (summarized in [75]). In a number of types of malignant tumors (oncocytomas), mitochondrial abnormalities are not only accompanied by uncontrolled cell proliferation, but also by inexplicably high mitochondrial proliferation, often resulting in mitochondria occupying the entire cytoplasm (Fig. 4). Note that oncocytes, in particular, appear as a toxic result of damage to mitochondria (Fig. 4b shows oncocytoma caused by lung hyperoxia, and Fig. 4c – by the effect of a liver carcinogen). The development of this type of cancer is believed to result from mDNA damage [80] leading to complex I damage [81]. There is a direct correlation between the low content of mitochondrial respiratory chain components and high level of tumor malignancy [82]. Given all these observations, the pathogenic role of mitochondrial ROS in oncogenesis becomes apparent [83, 84]. The final minimal statement includes a clear conclusion – accumulation of mitochondrial damage with time leads to the formation of a malignant tumor. It should be noted that normal mitochondrial functioning suppresses tumor formation [85]. This allows attributing to mitochondria the role of the supreme judge making a decision on whether the cell turns into a malignant one or not.
Fig. 4. Mitochondrial proliferation in oncocytes. a) Oncocytes in parotid gland with cytoplasm completely filled with mitochondria; arrows indicate mitochondrial inclusions ([128], reprinted with permission); b) oncocyte in contact with alveolar space (A) in lungs of newborn rats breathing 100% oxygen for six months; similar to (a), cytoplasm of an oncocyte surrounded by fibrous tissue is completely filled with proliferating mitochondria ([129], reprinted with permission); c) oncocyte in kidney epithelium of a rat receiving nitrosomorpholine for several weeks. Some mitochondria are organized in cylinders ([130], reprinted with permission).
KNOWN BACTERIAL AND POSSIBLE MITOCHONDRIAL INFECTION
While most bacteria occupy the extracellular phase and multiply there, some get into cells, determining the degree of bacterial invasion. Bacterial invasion was primarily detected for macrophages and epitheliocytes, and the method of bacterial penetration is different for these two cell types. Bacteria get into macrophages due to endocytosis, and for epithelial cells a set of factors is required – bacteria consecutively use these factors so that they finally enter the cytoplasm of the target cell. Bacterial invasion is known both for Gram-negative (e.g. Escherichia, Chlamydia, Klebsiella, Pseudomonas, Yersinia, Legionella, Staphylococcus, Salmonella) and Gram-positive bacteria (Listeria, Mycobacterium). Mechanisms of bacterial invasion of cells are different. For some bacteria, the initial stage of invasion is binding to integrins of the host cells, while others are engulfed by endocytosis without the stage of integrin-binding. Bacterial endocytosis is characteristic of epithelial cells and macrophages. After endocytic vacuoles with live bacteria get inside the cell, the vacuolar membrane is lysed and the bacteria are released into the cytoplasm, where they function exclusively as parasites.
Gram-negative bacteria produce toxins and enzymes that provide their binding with the host cell. In this way they realize the pathogenic effect without entering the cell. For example, all Gram-negative bacteria produce endotoxin, lipopolysaccharide (LPS), which is a component of cell walls [86-88]. According to one accepted model of sepsis, LPS plays the key role in this phenomenon, although this model is far from being perfect and is an object of constant criticism [89]. LPS injection into the bloodstream causes a number of pathological symptoms in a macroorganism: first, inflammatory response, with significant role of inflammatory mediators such as tumor necrosis factor and interleukin-1 [89]. In the most extreme cases a state of endotoxic shock accompanied by a massive cell death can be observed. This suggests that pathology of bacterial infection is largely determined by the effect of primary bacterial toxins and secondary factors mediated by the primary ones. Thus, bacteria do not necessarily need to enter a healthy cell to manifest their pathological effect. Instead, pathogenic exocellular factors, which cause damage or death of the host cell, can be used for that purpose.
Mitochondrial transfection, i.e. the possibility of alien mitochondria from the extracellular medium entering a cell, is discussed very rarely, simply because such a possibility seems to be rather absurd: extra- and intracellular conditions differ too much in their chemical composition, and it seems to exclude the presence of “free-living” mitochondria. However, there are a number of studies on mitochondria entering cells from neighboring contacting cells. The possibility of their transport along tunneling nanotubes (formations of the cytoplasmic membrane providing intercellular communication) is discussed, and we cannot exclude that such transport can involve gap junctions [90-93]. There is a trend to assume that this transfer of mitochondria could help in directing the differentiation of undifferentiated cells and provide the rescue of cells from mitochondrial defects [91, 92, 94]. In principle, such a transfer can have two results: transfer of “sick” mitochondria into a healthy cell or transfer of “healthy” mitochondria into a sick cell. In the first case, such a transfer would have a pathogenic, and in the second – healing effect, suggesting a possible dual effect of mitochondrial transfection. In earlier studies, the necessity of transferring the whole mitochondria was unclear, because, in principle, it was enough to transfer only mDNA to the defective cell to introduce new properties [95]. Cell transfection with mDNA is theoretically possible, and we are aware of many successful attempts to carry out this process [96]. Note that free mDNA is easily detected in blood, especially in patients after trauma [97].
One of the first demonstrations of the possibility to introduce new cellular properties by means of introduction of alien mitochondria to a cell was carried out in 1982. In experiments on co-cultivation of cells sensitive to chloramphenicol and efrapeptin, and mitochondria isolated from cells resistant to these drugs, the appearance of mitochondria in the cells (probably due to endocytosis) accompanied by the appearance of resistance of the cells to these antibiotics was observed [98].
We could draw a parallel between bacterial toxins (e.g. LPS) and already mentioned mitochondrial components (intact or modified [99]) DAMPs [100], which, similarly to bacterial toxins, bind to cell receptors represented by the family of Toll-like receptors [101], in particular, TLR-4 and TLR-9 [100, 102, 103], initiating the innate immune response.
MITOCHONDRIAL (OXIDATIVE) THEORY OF AGING AND BACTERIAL
INVOLVEMENT IN THIS PROCESS
In this review, we have not considered in detail the oxidative (mitochondrial) theory of aging [104, 105], since there are many excellent reviews on this topic [106-108]. Let us note only the essence of this theory, which states that oxidative damage determines the aging process and, ultimately, the lifespan. Given that mitochondria are perhaps the main producer of oxidative equivalents, it is mitochondria that determine the aging process. There are opponents of the mitochondrial theory of aging since not all the representatives of the animal kingdom logically follow the rules of this theory (see reviews [109-112]). However, the proponents of this theory are still in majority (a review clarifying different aspects of the oxidative theory of aging and providing explanation of the existing controversies can be found in [113]).
As we have discussed above, there is an assumption that bacteria inhabiting higher organisms determine their aging. We have also quoted the words by Metchnikoff, who claimed that human aging is determined by intestinal microbiota, for which we are the host.
Recently, a review titled “You are what you host: microbiome modulation of the aging process” was published in the journal Cell [114]; the review provides a detailed analysis of the regulation of the aging of a host by the bacteria inhabiting the host. Although aging of the nematode C. elegans is considered in that review, the basic ideas are still applicable to any host inhabited by microbiota. Two components of interspecies interaction of higher organisms and their bacteria are analyzed – symbiosis and pathogenesis. Taking into account the close spatial proximity of bacteria and their hosts, diffusible molecules produced by bacteria are able to directly affect host cells. A striking example is the suppression of OP50 E. coli strain, which is present in old nematodes and is considered nonpathogenic because it increases their lifespan [115].
Perhaps we are now at the beginning of a new stage of reconsideration of the role of mitochondria in the life of an organism. The previous revolutionary change in mitochondriology occurred around 1996, when we witnessed an abrupt transition from the traditional understanding of mitochondria purely as an energy machine to alternative mitochondrial functions, and above all, to the role of mitochondria in cell death. This revolution led to a substantial redistribution of resources and migration of a significant part of the biological research community, primarily molecular biologists, immunologists, and specialists in intracellular signalization, to mitochondriology. On one hand, it has strengthened general interest in mitochondria, accompanied by an increase in mitochondrial research funding, but on the other hand, the new researchers flooded this area with their previous ideological and practical luggage. As a result, the old solid studies on the structure of mitochondria and their correlation with cellular functions were forgotten. The purpose of this review is educational, above all, for nonclassical mitochondriologists to help them look at mitochondria from a slightly different angle, taking into account its possible bacterial origin. If such an approach makes sense, we need to mobilize the microbiological arsenal, in particular, to address the issue we have just touched. If we assume that microbiota, inhabiting only part of the macroorganism, determines most of the pathologies inherent to the host, including the organization of its aging and death, then there appears a valid question: Can mitobiota inhabiting all cells of the host also determine the same processes? To stimulate such considerations, we have presented an outline of data supporting the possibility and validity of such comparisons.
This work was supported by the Russian Foundation for Basic Research (grants 13-04-00484, 14-04-00300, 14-04-00542), the Russian Science Foundation (grant 14-24-00107), and the Program of the President of the Russian Federation (MK-2508.2014.4).
REFERENCES
1.Zorov, D. B., Plotnikov, E. Y., Jankauskas, S. S.,
Isaev, N. K., Silachev, D. N., Zorova, L. D., Pevzner, I. B., Pulkova,
N. V., Zorov, S. D., and Morosanova, M. A. (2012) The phenoptosis
problem: what is causing the death of an organism? Lessons from acute
kidney injury, Biochemistry (Moscow), 77, 742-753.
2.Zorov, D. B., Isaev, N. K., Plotnikov, E. Y.,
Silachev, D. N., Zorova, L. D., Pevzner, I. B., Morosanova, M. A.,
Jankauskas, S. S., Zorov, S. D., and Babenko, V. A. (2013) Perspectives
of mitochondrial medicine, Biochemistry (Moscow), 78,
979-990.
3.Henle, J. (1841) Mischungs und forbestandteile des
menschlichen korpers, in Allgemeine Anatomie, Varlag Leopold
Voss, Leipzig, pp. 573-613.
4.Kolliker, A. (1857) Einige bemerkungen uber die
endigungen der hautnerven und den bau der muskein, Zwiss Zool.,
8, 311-325.
5.Retzius, G. (1890) Muskelfibrille und sarcoplasma,
Biol. Untersuchungen Neue Folge, 1, 51-88.
6.Cajal, S. (1888) Observations sur la texture des
fibres musculaires des pattes et des ailes des insects, Int.
Monatszeitschrift Anat. Physiol., 205-232, 253-276.
7.Regaud, C., and Favre, M. (1909) Granulations
interstitielles et mitochondries des fibres musculaires striees,
Compt. Rend., 148, 661-664.
8.Benda, C. (1900) Weitere beobachtungen uber die
mitochondria und ihr verhaltnus zu sekretgranulationen nebst kritischen
bemerkungen, Arch. Anat. Physiol., 24, 166-178.
9.Mereschkowski, C. (1905) Uber natur und ursprung
der chromatophoren im pflanzenreiche, Biol. Centralbl.,
25, 593-604.
10.Mereschkowsky, K. (1910) Theorie der zwei
plasmaarten als grundlage der symbiogenesis, einer neuen lehre von der
ent-stehung der organismen, Biol. Centralbl., 30,
353-367.
11.Schimper, A. (1883) Uber die entwicklung der
chlorophyllkorner und farbkorper, Bot. Zeitung, 30,
105-114, 121-131, 137-146, 153-162.
12.Wallin, I. (1923) The mitochondria problem,
Amer. Nat., 57, 255-261.
13.Wallin, I. (1927) Symbionticism and the origin of
species, in The American Naturalist, Williams & Wilkins
Company, Baltimore.
14.Sagan, L. (1967) On the origin of mitosing cells,
J. Theor. Biol., 14, 255-274.
15.Margulis, L. (1971) Symbiosis and evolution,
Sci. Am., 225, 48-57.
16.Harada, S., Inaoka, D. K., Ohmori, J., and Kita,
K. (2013) Diversity of parasite complex II, Biochim. Biophys.
Acta, 1827, 658-667.
17.Collins, T. J., Berridge, M. J., Lipp, P., and
Bootman, M. D. (2002) Mitochondria are morphologically and functionally
heterogeneous within cells, EMBO J., 21, 1616-1627.
18.Sutovsky, P., Moreno, R. D., Ramalho-Santos, J.,
Dominko, T., Simerly, C., and Schatten, G. (1999) Ubiquitin tag for
sperm mitochondria, Nature, 402, 371-372.
19.Jin, S. M., Lazarou, M., Wang, C., Kane, L. A.,
Narendra, D. P., and Youle, R. J. (2010) Mitochondrial membrane
potential regulates PINK1 import and proteolytic destabilization by
PARL, J. Cell Biol., 191, 933-942.
20.Hawrelak, J. A., and Myers, S. P. (2004) The
causes of intestinal dysbiosis: a review, Altern. Med.
Rev., 9, 180-197.
21.Kuhne, L. (1893) Die neue Heilwissenschaft
oder die Lehre von der Einheit aller Krankheiten und deren darauf
begrundete einheitliche, arzneilose und operationslose
Heilung, Verlag von Louis Kuhne, Leipzig.
22.Mechnikov, I. (1915) On the Nature of Man
[in Russian], Nauchnoe Slovo.
23.Dominguez, J. A., and Coopersmith, C. M. (2010)
Can we protect the gut in critical illness? The role of growth factors
and other novel approaches, Crit. Care Clin., 26,
549-565.
24.Schwartz, R. F., Neu, J., Schatz, D., Atkinson,
M. A., and Wasserfall, C. (2007) Comment on: Brugman, S., et al. (2006)
Antibiotic treatment partially protects against type 1 diabetes in the
bio-breeding diabetes-prone rat. Is the gut flora involved in the
development of type 1 diabetes? Diabetologia, 50,
220-221.
25.Cani, P. D., Bibiloni, R., Knauf, C., Waget, A.,
Neyrinck, A. M., Delzenne, N. M., and Burcelin, R. (2008) Changes in
gut microbiota control metabolic endotoxemia-induced inflammation in
high-fat-diet-induced obesity and diabetes in mice, Diabetes,
57, 1470-1481.
26.Cani, P. D., Neyrinck, A. M., Fava, F., Knauf,
C., Burcelin, R. G., Tuohy, K. M., Gibson, G. R., and Delzenne, N. M.
(2007) Selective increases of bifidobacteria in gut microflora improve
high-fat-diet-induced diabetes in mice through a mechanism associated
with endotoxemia, Diabetologia, 50, 2374-2383.
27.Bukowska, H., Pieczul-Mroz, J., Jastrzebska, M.,
Chelstowski, K., and Naruszewicz, M. (1998) Decrease in fibrinogen and
LDL-cholesterol levels upon supplementation of diet with
Lactobacillus plantarum in subjects with moderately elevated
cholesterol, Atherosclerosis, 137, 437-438.
28.Shimizu, K., Ogura, H., Goto, M., Asahara, T.,
Nomoto, K., Morotomi, M., Yoshiya, K., Matsushima, A., Sumi, Y.,
Kuwagata, Y., Tanaka, H., Shimazu, T., and Sugimoto, H. (2006) Altered
gut flora and environment in patients with severe SIRS, J.
Trauma, 60, 126-133.
29.Maejima, K., Deitch, E., and Berg, R. (1984)
Promotion by burn stress of the translocation of bacteria from the
gastrointestinal tracts of mice, Arch. Surg., 119,
166-172.
30.Maejima, K., Deitch, E. A., and Berg, R. D.
(1984) Bacterial translocation from the gastrointestinal tracts of rats
receiving thermal injury, Infect. Immun., 43, 6-10.
31.LeVoyer, T., Cioffi, W. G., Jr., Pratt, L.,
Shippee, R., McManus, W. F., Mason, A. D., Jr., and Pruitt, B. A., Jr.
(1992) Alterations in intestinal permeability after thermal injury,
Arch. Surg., 127, 26-29, discussion 29-30.
32.Bolte, E. R. (1998) Autism and Clostridium
tetani, Med. Hypotheses, 51, 133-144.
33.Finegold, S. M., Molitoris, D., Song, Y., Liu,
C., Vaisanen, M. L., Bolte, E., McTeague, M., Sandler, R., Wexler, H.,
Marlowe, E. M., Collins, M. D., Lawson, P. A., Summanen, P., Baysallar,
M., Tomzynski, T. J., Read, E., Johnson, E., Rolfe, R., Nasir, P.,
Shah, H., Haake, D. A., Manning, P., and Kaul, A. (2002)
Gastrointestinal microflora studies in late-onset autism, Clin.
Infect. Dis., 35, S6-S16.
34.McKeever, T. M., Lewis, S. A., Smith, C.,
Collins, J., Heatlie, H., Frischer, M., and Hubbard, R. (2002) Early
exposure to infections and antibiotics and the incidence of allergic
disease: a birth cohort study with the West Midlands General Practice
Research Database, J. Allergy Clin. Immunol., 109,
43-50.
35.Sekirov, I., Russell, S. L., Antunes, L. C., and
Finlay, B. B. (2010) Gut microbiota in health and disease, Physiol.
Rev., 90, 859-904.
36.Angus, D. C. (2011) The search for effective
therapy for sepsis: back to the drawing board? J. Am. Med.
Assoc., 306, 2614-2615.
37.Cauwels, A., Rogge, E., Vandendriessche, B.,
Shiva, S., and Brouckaert, P. (2014) Extracellular ATP drives systemic
inflammation, tissue damage and mortality, Cell Death Dis.,
5, e1102.
38.Skulachev, V. P. (2002) Programmed death
phenomena: from organelle to organism, Ann. NY Acad. Sci.,
959, 214-237.
39.Agteresch, H. J., Dagnelie, P. C., van den Berg,
J. W., and Wilson, J. H. (1999) Adenosine triphosphate: established and
potential clinical applications, Drugs, 58, 211-232.
40.Murry, C. E., Jennings, R. B., and Reimer, K. A.
(1986) Preconditioning with ischemia: a delay of lethal cell injury in
ischemic myocardium, Circulation, 74, 1124-1136.
41.LeBlanc, J., Roberge, C., Valliere, J., and
Oakson, G. (1971) The sympathetic nervous system in short-term
adaptation to cold, Can. J. Physiol. Pharmacol., 49,
96-101.
42.Yun, J., and Finkel, T. (2014) Mitohormesis,
Cell Metab., 19, 757-766.
43.Skulachev, V. P. (1996) Role of uncoupled and
non-coupled oxidations in maintenance of safely low levels of oxygen
and its one-electron reductants, Q. Rev. Biophys., 29,
169-202.
44.Cunha, F. M., Caldeira da Silva, C. C.,
Cerqueira, F. M., and Kowaltowski, A. J. (2011) Mild mitochondrial
uncoupling as a therapeutic strategy, Curr. Drug Targets,
12, 783-789.
45.Abbracchio, M. P., Burnstock, G., Verkhratsky,
A., and Zimmermann, H. (2009) Purinergic signalling in the nervous
system: an overview, Trends Neurosci., 32, 19-29.
46.Krysko, O., Love Aaes, T., Bachert, C.,
Vandenabeele, P., and Krysko, D. V. (2013) Many faces of DAMPs in
cancer therapy, Cell Death Dis., 4, e631.
47.Boya, P., Roques, B., and Kroemer, G. (2001) New
EMBO members’ review: viral and bacterial proteins regulating
apoptosis at the mitochondrial level, EMBO J., 20,
4325-4331.
48.Kozjak-Pavlovic, V., Ross, K., and Rudel, T.
(2008) Import of bacterial pathogenicity factors into mitochondria,
Curr. Opin. Microbiol., 11, 9-14.
49.Kroemer, G., Galluzzi, L., and Brenner, C. (2007)
Mitochondrial membrane permeabilization in cell death, Physiol.
Rev., 87, 99-163.
50.Skulachev, V. P. (2000) Mitochondria in the
programmed death phenomena; a principle of biology: “it is better
to die than to be wrong”, Life, 49, 365-373.
51.Macbride, D. (1772) A Methodical Introduction
to the Theory and Practice of Physic, Science.
52.Onuigbo, W. I. (1975) Some nineteenth century
ideas on links between tuberculous and cancerous diseases of the lung,
Br. J. Dis. Chest, 69, 207-210.
53.Broxmeyer, L. (2004) Is cancer just an incurable
infectious disease? Med. Hypotheses, 63, 986-996.
54.Virchow, R. (1860) Cellular Pathology,
Churchill, London.
55.Virchow, R. (1863) Die Krankhaften
Geschwulste, August Hirshwald, Berlin.
56.Balkwill, F., and Mantovani, A. (2001)
Inflammation and cancer: back to Virchow? Lancet, 357,
539-545.
57.Morrison, W. B. (2012) Inflammation and cancer: a
comparative view, J. Vet. Intern. Med., 26, 18-31.
58.Bierne, H., Hamon, M., and Cossart, P. (2014)
Epigenetics and bacterial infections, Cold Spring Harb. Perspect.
Med., 2, a010272.
59.Gaylord, H. R. (1901) The protozoon of cancer. A
preliminary report based upon three years’ work in the New York
State Pathological Laboratory of the University of Buffalo, Am. J.
Med. Sci., 121, 503-539.
60.Wainwright, A. M. (2006) The potential role of
non-virus microorganisms in cancer, Curr. Trends Microbiol.,
48-59.
61.Peter, S., and Beglinger, C. (2007)
Helicobacter pylori and gastric cancer: the causal relationship,
Digestion, 75, 25-35.
62.Correa, P., and Houghton, J. (2007)
Carcinogenesis of Helicobacter pylori, Gastroenterology,
133, 659-672.
63.Hussell, T., Isaacson, P. G., Crabtree, J. E.,
and Spencer, J. (1993) The response of cells from low-grade B-cell
gastric lymphomas of mucosa-associated lymphoid tissue to
Helicobacter pylori, Lancet, 342, 571-574.
64.Mc, C. W., and Mason, J. M., 3rd (1951)
Enterococcal endocarditis associated with carcinoma of the sigmoid:
report of a case, J. Med. Assoc. State Ala., 21,
162-166.
65.Littman, A. J., Jackson, L. A., and Vaughan, T.
L. (2005) Chlamydia pneumoniae and lung cancer: epidemiological
evidence, Cancer Epidemiol. Biomarkers Prev., 14,
773-778.
66.Littman, A. J., White, E., Jackson, L. A.,
Thornquist, M. D., Gaydos, C. A., Goodman, G. E., and Vaughan, T. L.
(2004) Chlamydia pneumoniae infection and risk of lung cancer,
Cancer Epidemiol. Biomarkers Prevent., 13, 1624-1630.
67.Kovalchuk, O., Walz, P., and Kovalchuk, I. (2014)
Does bacterial infection cause genome instability and cancer in the
host cell? Mutat. Res., 761C, 1-14.
68.Parsonnet, J. (1995) Bacterial infection as a
cause of cancer, Environ. Health Perspect., 103, Suppl.
8, 263-268.
69.Szent-Gyorgyi, A. (1977) The living state and
cancer, Proc. Natl. Acad. Sci. USA, 74, 2844-2847.
70.Warburg, O. (1956) On the origin of cancer cells,
Science, 123, 309-314.
71.Zorov, D. B. (1996) Mitochondrial damage as a
source of diseases and aging: a strategy of how to fight these,
Biochim. Biophys. Acta, 1275, 10-15.
72.Zorov, D. B., Krasnikov, B. F., Kuzminova, A. E.,
Vysokikh, M., and Zorova, L. D. (1997) Mitochondria revisited.
Alternative functions of mitochondria, Biosci. Rep., 17,
507-520.
73.Ramanathan, A., Wang, C., and Schreiber, S. L.
(2005) Perturbational profiling of a cell-line model of tumorigenesis
by using metabolic measurements, Proc. Natl. Acad. Sci. USA,
102, 5992-5997.
74.Mayevsky, A. (2009) Mitochondrial function and
energy metabolism in cancer cells: past overview and future
perspectives, Mitochondrion, 9, 165-179.
75.Seyfried, T. N., and Shelton, L. M. (2010) Cancer
as a metabolic disease, Nutr. Metab. (Lond.), 7, 7.
76.Roskelley, R. C., Mayer, N., Horwitt, B. N., and
Salter, W. T. (1943) Studies in cancer. VII. Enzyme deficiency in human
and experimental cancer, J. Clin. Invest., 22,
743-751.
77.John, A. P. (2001) Dysfunctional mitochondria,
not oxygen insufficiency, cause cancer cells to produce inordinate
amounts of lactic acid: the impact of this on the treatment of cancer,
Med. Hypotheses, 57, 429-431.
78.Galluzzi, L., Morselli, E., Kepp, O., Vitale, I.,
Rigoni, A., Vacchelli, E., Michaud, M., Zischka, H., Castedo, M., and
Kroemer, G. (2010) Mitochondrial gateways to cancer, Mol. Aspects
Med., 31, 1-20.
79.Cuezva, J. M., Krajewska, M., de Heredia, M. L.,
Krajewski, S., Santamaria, G., Kim, H., Zapata, J. M., Marusawa, H.,
Chamorro, M., and Reed, J. C. (2002) The bioenergetic signature of
cancer: a marker of tumor progression, Cancer Res., 62,
6674-6681.
80.Welter, C., Kovacs, G., Seitz, G., and Blin, N.
(1989) Alteration of mitochondrial DNA in human oncocytomas, Genes
Chromosomes Cancer, 1, 79-82.
81.Savagner, F., Franc, B., Guyetant, S., Rodien,
P., Reynier, P., and Malthiery, Y. (2001) Defective mitochondrial ATP
synthesis in oxyphilic thyroid tumors, J. Clin. Endocrinol.
Metab., 86, 4920-4925.
82.Simonnet, H., Alazard, N., Pfeiffer, K., Gallou,
C., Beroud, C., Demont, J., Bouvier, R., Schagger, H., and Godinot, C.
(2002) Low mitochondrial respiratory chain content correlates with
tumor aggressiveness in renal cell carcinoma, Carcinogenesis,
23, 759-768.
83.Bonora, E., Porcelli, A. M., Gasparre, G.,
Biondi, A., Ghelli, A., Carelli, V., Baracca, A., Tallini, G.,
Martinuzzi, A., Lenaz, G., Rugolo, M., and Romeo, G. (2006) Defective
oxidative phosphorylation in thyroid oncocytic carcinoma is associated
with pathogenic mitochondrial DNA mutations affecting complexes I and
III, Cancer Res., 66, 6087-6096.
84.Demasi, A. P., Furuse, C., Altemani, A.,
Junqueira, J. L., Oliveira, P. R., and Araujo, V. C. (2009)
Peroxiredoxin I is overexpressed in oncocytic lesions of salivary
glands, J. Oral Pathol. Med., 38, 514-517.
85.Israel, B. A., and Schaeffer, W. I. (1987)
Cytoplasmic suppression of malignancy, In vitro Cell Dev. Biol.,
23, 627-632.
86.Raetz, C. R., and Whitfield, C. (2002)
Lipopolysaccharide endotoxins, Annu. Rev. Biochem., 71,
635-700.
87.Wang, X., and Quinn, P. J. (2010) Endotoxins:
lipopolysaccharides of gram-negative bacteria, Subcell.
Biochem., 53, 3-25.
88.Heumann, D., and Roger, T. (2002) Initial
responses to endotoxins and Gram-negative bacteria, Clin. Chim.
Acta, 323, 59-72.
89.Remick, D. G., and Ward, P. A. (2005) Evaluation
of endotoxin models for the study of sepsis, Shock, 24,
Suppl. 1, 7-11.
90.Koyanagi, M., Brandes, R. P., Haendeler, J.,
Zeiher, A. M., and Dimmeler, S. (2005) Cell-to-cell connection of
endothelial progenitor cells with cardiac myocytes by nanotubes: a
novel mechanism for cell fate changes? Circ. Res., 96,
1039-1041.
91.Plotnikov, E. Y., Khryapenkova, T. G., Galkina,
S. I., Sukhikh, G. T., and Zorov, D. B. (2010) Cytoplasm and organelle
transfer between mesenchymal multipotent stromal cells and renal
tubular cells in co-culture, Exp. Cell Res., 316,
2447-2455.
92.Plotnikov, E. Y., Pulkova, N. V., Pevzner, I. B.,
Zorova, L. D., Silachev, D. N., Morosanova, M. A., Sukhikh, G. T., and
Zorov, D. B. (2013) Inflammatory pre-conditioning of mesenchymal
multipotent stromal cells improves their immunomodulatory potency in
acute pyelonephritis in rats, Cytotherapy, 15,
679-689.
93.Prockop, D. J. (2012) Mitochondria to the rescue,
Nature Med., 18, 653-654.
94.Spees, J. L., Olson, S. D., Whitney, M. J., and
Prockop, D. J. (2006) Mitochondrial transfer between cells can rescue
aerobic respiration, Proc. Natl. Acad. Sci. USA, 103,
1283-1288.
95.Csordas, A. (2006) Mitochondrial transfer between
eukaryotic animal cells and its physiologic role, Rejuvenation
Res., 9, 450-454.
96.Mileshina, D., Ibrahim, N., Boesch, P.,
Lightowlers, R. N., Dietrich, A., and Weber-Lotfi, F. (2011)
Mitochondrial transfection for studying organellar DNA repair, genome
maintenance and aging, Mech. Ageing Dev., 132,
412-423.
97.Zhang, Q., Itagaki, K., and Hauser, C. J. (2010)
Mitochondrial DNA is released by shock and activates neutrophils via
p38 map kinase, Shock, 34, 55-59.
98.Clark, M. A., and Shay, J. W. (1982)
Mitochondrial transformation of mammalian cells, Nature,
295, 605-607.
99.Shimada, K., Crother, T. R., Karlin, J.,
Dagvadorj, J., Chiba, N., Chen, S., Ramanujan, V. K., Wolf, A. J.,
Vergnes, L., Ojcius, D. M., Rentsendorj, A., Vargas, M., Guerrero, C.,
Wang, Y., Fitzgerald, K. A., Underhill, D. M., Town, T., and Arditi, M.
(2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome
during apoptosis, Immunity, 36, 401-414.
100.Zhang, Q., Raoof, M., Chen, Y., Sumi, Y.,
Sursal, T., Junger, W., Brohi, K., Itagaki, K., and Hauser, C. J.
(2010) Circulating mitochondrial DAMPs cause inflammatory responses to
injury, Nature, 464, 104-107.
101.Medzhitov, R., Preston-Hurlburt, P., and
Janeway, C. A., Jr. (1997) A human homologue of the Drosophila
Toll protein signals activation of adaptive immunity, Nature,
388, 394-397.
102.Wright, S. D. (1999) Toll, a new piece in the
puzzle of innate immunity, J. Exp. Med., 189,
605-609.
103.Krysko, D. V., Agostinis, P., Krysko, O., Garg,
A. D., Bachert, C., Lambrecht, B. N., and Vandenabeele, P. (2011)
Emerging role of damage-associated molecular patterns derived from
mitochondria in inflammation, Trends Immunol., 32,
157-164.
104.Harman, D. (1956) Aging: a theory based on free
radical and radiation chemistry, J. Gerontol., 11,
298-300.
105.Harman, D. (1992) Free radical theory of aging,
Mutat. Res., 275, 257-266.
106.Vina, J., Borras, C., Abdelaziz, K. M.,
Garcia-Valles, R., and Gomez-Cabrera, M. C. (2013) The free radical
theory of aging revisited: the cell signaling disruption theory of
aging, Antioxid. Redox Signal., 19, 779-787.
107.Barja, G. (2013) Updating the mitochondrial
free radical theory of aging: an integrated view, key aspects, and
confounding concepts, Antioxid. Redox Signal., 19,
1420-1445.
108.Liochev, S. I. (2013) Reactive oxygen species
and the free radical theory of aging, Free Radic. Biol. Med.,
60, 1-4.
109.Perez, V. I., Bokov, A., Van Remmen, H., Mele,
J., Ran, Q., Ikeno, Y., and Richardson, A. (2009) Is the oxidative
stress theory of aging dead? Biochim. Biophys. Acta,
1790, 1005-1014.
110.Lapointe, J., and Hekimi, S. (2010) When a
theory of aging ages badly, Cell Mol. Life Sci., 67,
1-8.
111.Speakman, J. R., and Selman, C. (2011) The
free-radical damage theory: accumulating evidence against a simple link
of oxidative stress to ageing and lifespan, BioEssays: News Rev.
Mol. Cell. Devel. Biol., 33, 255-259.
112.Gladyshev, V. N. (2014) The free radical theory
of aging is dead. Long live the damage theory! Antioxid. Redox
Signal., 20, 727-731.
113.Kirkwood, T. B., and Kowald, A. (2012) The
free-radical theory of ageing – older, wiser and still alive:
modelling positional effects of the primary targets of ROS reveals new
support, BioEssays: News Rev. Mol. Cell. Devel. Biol.,
34, 692-700.
114.Heintz, C., and Mair, W. (2014) You are what
you host: microbiome modulation of the aging process, Cell,
156, 408-411.
115.Zhang, R., and Hou, A. (2013)
Host–microbe interactions in Caenorhabditis elegans,
ISRN Microbiol., DOI.10.1155/2013/356451.
116.Bakeeva, L. E., Chentsov Yu. S., and Skulachev,
V. P. (1983) Intermitochondrial contacts in myocardiocytes, J. Mol.
Cell Cardiol., 15, 413-420.
117.Suzuki, T., and Mostofi, F. K. (1967)
Intramitochondrial filamentous bodies in the thick limb of henle of the
rat kidney, J. Cell Biol., 33, 605-623.
118.Sarnat, H. B., Flores-Sarnat, L., Casey, R.,
Scott, P., and Khan, A. (2012) Endothelial ultrastructural alterations
of intramuscular capillaries in infantile mitochondrial cytopathies:
“mitochondrial angiopathy”, Neuropathology,
32, 617-627.
119.Hawkins, W. E., Howse, H. D., and Foster, C. A.
(1980) Prismatic cristae and paracrystalline inclusions in mitochondria
of myocardial cells of the oyster Crassostrea virginica Gmelin,
Cell Tissue Res., 209, 87-94.
120.Behbehani, A. W., Goebel, H., Osse, G.,
Gabriel, M., Langenbeck, U., Berden, J., Berger, R., and Schutgens, R.
B. (1984) Mitochondrial myopathy with lactic acidosis and deficient
activity of muscle succinate cytochrome-c-oxidoreductase,
Eur. J. Pediatr., 143, 67-71.
121.Buell, R., Wang, N. S., Seemayer, T. A., and
Ahmed, M. N. (1976) Endobronchial plasma cell granuloma (xanthomatous
pseudotumor); a light and electron microscopic study, Hum.
Pathol., 7, 411-426.
122.Blinzinger, K., Rewcastle, N. B., and Hager, H.
(1965) Observations on prismatic-type mitochondria within astrocytes of
the Syrian hamster brain, J. Cell Biol., 25, 293-303.
123.Vital, A., and Vital, C. (2012) Mitochondria
and peripheral neuropathies, J. Neuropathol. Exp. Neurol.,
71, 1036-1046.
124.Van Ekeren, G. J., Stadhouders, A. M.,
Egberink, G. J., Sengers, R. C., Daniels, O., and Kubat, K. (1987)
Hereditary mitochondrial hypertrophic cardiomyopathy with mitochondrial
myopathy of skeletal muscle, congenital cataract and lactic acidosis,
Virchows Arch. Pathol. Anat. Histopathol., 412,
47-52.
125.Andersson-Cedergren, E. (1959) Ultrastructure
of motor end plate and sarcoplasmic components of mouse skeletal muscle
fiber as revealed by three-dimensional reconstructions from serial
sections, J. Ultrastruct. Res., 2, Suppl. 1, 5-191.
126.Mannella, C. A., Marko, M., and Buttle, K.
(1997) Reconsidering mitochondrial structure: new views of an old
organelle, TIBS, 22, 37-38.
127.Daems, W. T., and Wisse, E. (1966) Shape and
attachment of the cristae mitochondriales in mouse hepatic cell
mitochondria, J. Ultrastruct. Res., 16, 123-140.
128.Sun, C. N., White, H. J., and Thompson, B. W.
(1975) Oncocytoma (mitochondrioma) of the parotid gland. An electron
microscopical study, Arch. Pathol., 99, 208-214.
129.Bannasch, P., Krech, R., and Zerban, H. (1978)
Morphogenese und micromorphologie epithelialer nierentumoren bei
nitrosomorpholin-vergifteten ratten. III. Oncocytentubuli und
oncocytomas, Zeitschrift Krebsforschung Klin. Onkol. (Cancer Res.
Clin. Oncol.), 92, 87-104.
130.Bonikos, D. S., Bensch, K. G., Watt, T., and
Northway, W. H. (1977) Pulmonary oncocytes in prolonged hyperoxia,
Exp. Mol. Pathol., 26, 92-102.