Escherichia coli Signal Peptidase Recognizes and Cleaves Archaeal Signal Sequence
Majida Atta Muhammad1#, Samia Falak1#, Naeem Rashid1*, Qurra-tul-Ann Afza Gardner1, Nasir Ahmad2, Tadayuki Imanaka3, and Muhammad Akhtar1,4
1University of the Punjab, School of Biological Sciences, 54590 Lahore, Pakistan; E-mail: naeem.ff.sbs@pu.edu.pk, naeemrashid37@hotmail.com2University of the Punjab, Institute of Agricultural Sciences, 54590 Lahore, Pakistan
3Ritsumeikan University, The Research Organization of Science and Technology, 525-8577 Kusatsu, Shiga, Japan
4University of Southampton, School of Biological Sciences, Southampton SO16 7PX, UK
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received March 7, 2017; Revision received April 12, 2017
Tk1884, an open reading frame encoding α-amylase in Thermococcus kodakarensis, was cloned with the native signal sequence and expressed in Escherichia coli. Heterologous gene expression resulted in secretion of the recombinant protein to the extracellular culture medium. Extracellular α-amylase activity gradually increased after induction. Tk1884 was purified from the extracellular medium, and its molecular mass determined by electrospray ionization mass spectrometry indicated the cleavage of a few amino acids. The N-terminal amino acid sequence of the purified Tk1884 was determined, which revealed that the signal peptide was cleaved between Ala26 and Ala27 by E. coli signal peptidase. To the best of our knowledge, this is the first report describing an archaeal signal sequence recognized and cleaved by E. coli signal peptidase.
KEY WORDS: α-amylase, Thermococcus kodakarensis, signal peptide, purification, mass spectrometry, N-terminal sequencingDOI: 10.1134/S0006297917070070
In all the three domains of life, proteins that are destined to be transported across the membrane are usually produced with an intrinsic N-terminal sequence that must be removed on the trans-side of the membrane. This N-terminal sequence is known as a signal or leader peptide [1, 2]. Signal peptides are generally 20-30 amino acids (a.a.) long [3]. Although the structure of the cell membrane and cell wall of bacteria and archaea are significantly different, signal peptides from both origins consist of three distinct regions; an n-region that is followed by a hydrophobic core (h-region) and a carboxyl terminal region (c-region) containing a signal peptidase cleavage site [4, 5]. Like bacteria and eukarya, the archaeal signal peptide cleavage site follows the –3, –1 rule, in which these sites are occupied by small and neutral amino acids (usually Ala) [6].
Among bacteria, Escherichia coli is the most popular organism for production of recombinant proteins [7]. However, production of recombinant proteins in E. coli sometimes results in accumulation of improperly folded and inactive protein aggregates [8-10]. This can be avoided if the recombinant protein secretes outside the cell into the culture medium. This secretory production is advantageous in purification, minimizing protease attack, no methionine extension at the N-terminus, and substantial possibility of proper folding of the protein [11-13]. We previously showed that the E. coli secretion system not only recognizes the signal sequence of gram-positive bacteria including xylanase from Bacillus subtilis and α-amylase from Bacillus licheniformis, but also secretes the mature protein to the culture medium [14, 15]. In the present study, we have expressed in E. coli an α-amylase gene, Tk1884, from the hyperthermophilic archaeon Thermococcus kodakarensis, previously known as Pyrococcus sp. KOD1 [16, 17], with the native signal sequence, and shown that E. coli signal peptidases recognize and cleave the signal peptide and secrete the mature protein into the culture medium.
MATERIALS AND METHODS
Strains and plasmids. The hyperthermophilic archaeal strain T. kodakarensis KOD1 was used as a source of α-amylase gene, Tk1884. Escherichia coli DH5α was used for gene cloning and BL21-CodonPlus(DE3)-RIL (Stratagene, USA) for expression of the gene. Plasmids pTZ57R/T (Thermo Fisher Scientific, USA) and pET-21a(+) (Stratagene) were used for cloning and expression purposes, respectively.
Cloning and expression of Tk1884 gene. Tk1884 (accession No. WP_011250835) contains a native N-terminal signal sequence. This gene with signal sequence was amplified by polymerase chain reaction (PCR) using a set of forward (5′-CATATGAAGAAGTTTGTCGCCCTGCTC-3′) and reverse (5′-TCATCCAACCCCGCAGTAGCTC-3′) primers. The PCR-amplified DNA fragment was inserted into pTZ57R/T cloning vector. The resultant vector was named pTZ-Tk1884. For expression, the NdeI-BamHI gene fragment was liberated from pTZ-Tk1884 and inserted into pET-21a(+) expression vector digested with the same set of restriction enzymes. The resulting plasmid pET-Tk1884 was used to transform E. coli BL21-CodonPlus(DE3)-RIL cells. Host cells carrying pET-Tk1884 vector were grown overnight at 37°C in LB medium containing ampicillin (100 µg/ml). Fresh LB medium (500 ml) containing ampicillin (100 µg/ml) was used to dilute the culture (1%). Cultivation was continued until A660 reached 0.4. Heterologous gene expression was induced by the addition of 0.2 mM isopropyl-β-D-thiogalactopyranoside (IPTG), and incubation was continued at 37°C for 4, 8, and 20 h.
Purification of recombinant Tk1884. For purification of extracellular Tk1884, the culture broth of E. coli BL21-CodonPlus(DE3)-RIL cells containing pET-Tk1884 was centrifuged at 14,000g for 20 min, and the supernatant was filtered through MF-Millipore Type HAWP 0.45 µm filter assemblies. This supernatant was brought to 80% ammonium sulfate saturation and placed at 4°C overnight for the precipitation of extracellular proteins. The precipitates were collected by centrifugation at 14,000g for 30 min and dissolved in 50 mM Tris-HCl (pH 8.0). The recombinant Tk1884 was purified by hydrophobic column chromatography using an AKTA purifier chromatography system (GE Healthcare, Sweden). A HiTrap Phenyl HP (5 ml) column was equilibrated with 50 mM Tris-HCl (pH 8.0), and Tk1884 was applied to the column. The column was washed with the same buffer, and proteins bound to the column were eluted with a linear gradient of 50-1 mM Tris-HCl (pH 8.0). Eluted fractions were analyzed by SDS-PAGE, and fractions containing α-amylase activity were pooled, dialyzed against 50 mM Tris-HCl (pH 8.0), and further purified by anion-exchange column chromatography. A HiTrap QFF column was equilibrated with 50 mM Tris-HCl (pH 8.0), and dialyzed fractions after HiTrap Phenyl HP were applied to the column. The column was washed with the same buffer, and proteins bound to the column were eluted with a linear gradient of 0-1.5 M NaCl.
Enzyme activity assay. The quantitative assay for starch hydrolyzing activity was based on the measurements of reducing sugars by the dinitrosalicylic acid (DNS) method. The properly diluted Tk1884 (5 µl) was incubated at 95°C for 3 min with preincubated 195 µl of 1% soluble starch solution; the reaction was terminated by quenching on ice. This solution was then mixed with one volume of DNS solution (1% 3,5-dinitrosalicylic acid, 0.4 M NaOH and 30% sodium potassium tartrate). The solution was then heated on a boiling water bath for 5 min, cooled at room temperature, and the absorbance of the solution was measured at 540 nm. A control experiment containing all the above reagents except the enzyme was run at the same time.
Molecular mass determination. To determine the molecular mass by electrospray ionization mass spectrometry, the purified recombinant Tk1884 was acidified with formic acid and passed through spin column (Amersham). Analysis of the purified recombinant Tk1884 (2-3 μg/μl) was then performed on 6224 TOF LC/MS (Agilent Technologies, USA) by injecting 10 μl of the desalted sample. The data were acquired on MassHunter Workstation using ESI voltage of 3.5 kV, gas temperature 325°C, fragmentor and skimmer voltage 175 and 65 V, respectively, with a drying gas flow of 5 liters/min, and a nebulizer pressure of 30 psig. The online separation was performed on the reverse phase RP-HPLC column (Agilent Poroshell 300SB-C-18) at a flow rate of 0.2 ml/min, using a gradient of 2-60% of 100% acetonitrile in 0.1% formic acid (solvent B) with 0.1% formic acid water (solvent A). The data acquired were then processed for deconvolution using optimized maximum entropy algorithms.
N-terminal amino acid sequencing. The N-terminal amino acid residues of purified recombinant Tk1884 were determined commercially by AltaBioscience (UK).
RESULTS AND DISCUSSION
Production of Tk1884 in E. coli. We cloned and expressed in E. coli Tk1884 from T. kodakarensis with its native signal peptide (Fig. S1; see Supplement to this paper on the site of the journal (http://protein.bio.msu.ru/biokhimiya) and Springer site (Link.springer.com)). Most of the recombinant Tk1884 was produced in the insoluble form when host cells were induced with 0.2 mM IPTG at 37°C. However, a significant amount of the recombinant protein was produced in the soluble form when E. coli cells were cultivated at 17°C after induction. In addition to the intracellular activity, significantly high enzyme activity was also observed in the extracellular growth medium, which gradually increased with time after induction.
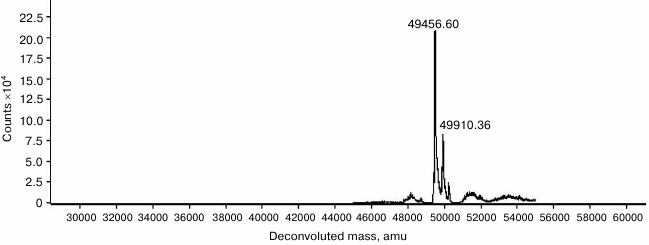
Purification, molecular mass determination, and N-terminal sequencing. Recombinant Tk1884 from the extracellular medium was purified to apparent homogeneity by ammonium sulfate precipitation and hydrophobic-interaction and ion-exchange column chromatographies (Fig. 1). The calculated molecular mass of Tk1884, based on amino acid sequence with signal sequence, was 52,213 Da. However, when the purified recombinant Tk1884 was subjected to SDS-PAGE analysis, it appeared at a molecular weight slightly lower than the theoretical one. Analysis of the amino acid sequence of Tk1884 for the presence of a signal peptide using a signal peptide prediction program (http://www.cbs.dtu.dk/services/SignalP/) for gram negative bacteria showed the presence of a signal peptide of 21 a.a.; the predicted signal peptide ended at Ala21 with a cleavage site between Ala21 and Gln22. However, for gram-positive bacteria and eukaryotes, the software predicted a signal peptide of 26 a.a. with a cleavage site between Ala26 and Ala27. Electrospray ionization mass spectrometry analysis of the Tk1884 purified from extracellular medium demonstrated two peaks, a major peak corresponding to 49,456 Da and a minor peak equivalent to 49,910 Da (Fig. 2). The calculated molecular weight of the mature protein, after cleavage of 21 a.a., was 49,913 Da, whereas cleavage of the signal peptide after 26 a.a. resulted in 49,459 Da. These calculated masses matched the masses of the two species of recombinant Tk1884 determined experimentally by electrospray ionization mass spectrometry (Table S1; see Supplement). This indicated that although the Tk1884 was cloned with signal sequence, the host E. coli cleaved the signal peptide releasing the mature Tk1884. To validate this, the purified recombinant Tk1884 was analyzed for the N-terminal amino acid sequence, and the following five amino acids were determined at the N-terminal of the purified protein: AKYSE (Fig. S2; see Supplement). These amino acid residues matched exactly the Tk1884 sequence after the 26th amino acid. This finding confirmed that the E. coli signal peptidase recognized the signal sequence of archaeal origin and cleaved it between Ala26 and Ala27.
Fig. 1. Coomassie brilliant blue stained SDS-PAGE demonstrating purified extracellular Tk1884. Lanes: 1) extracellular protein fraction of control (cells carrying pET-21a(+) vector); 2) extracellular protein fraction of cells carrying pET-Tk1884; 3) purified Tk1884 from extracellular medium.
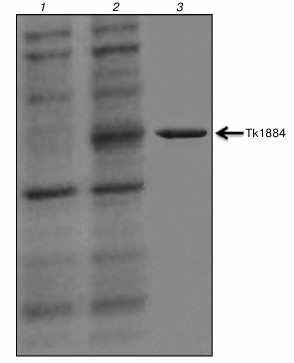
Fig. 2. Electrospray ionization mass spectrometry analysis of Tk1884 purified from the extracellular culture medium.
Levels of the α-amylase activity in different fractions. We further optimized the secretory expression by varying the time intervals after induction. The extracellular α-amylase activity increased, while the intracellular activity decreased with the passage of time. The extracellular and intracellular activities were 16% (4920 U/liter of the culture) and 84% (24,653 U/liter of the culture), respectively, after 4 h of induction, which changed to 35% (10,626 U/liter of the culture) and 65% (19,240 U/liter of the culture), respectively, after 8 h. The extracellular activity reached to 65% (19,190 U/liter of the culture) at 20 h post-induction. The characteristics of the outer membrane structure define the transfer of proteins after a polypeptide passes through the plasma membrane. Recombinant protein appears in a culture medium either by protein secretion by cells or disruption of cells during cultivation. To examine whether the extracellular α-amylase activity is a result of cell disruption or translocation of mature protein through the outer membrane of the cell after cleavage by signal peptidase, we measured the β-galactosidase activity in the extracellular medium. No β-galactosidase activity could be detected in the extracellular medium, indicating that the α-amylase activity in the extracellular medium was not due to the lysis of the cells. We further examined the levels of the α-amylase activity in the extracellular medium as well as in cytoplasmic and membrane fractions of E. coli cells. The soluble fraction after centrifugation at 14,000g for 20 min was centrifuged at 138,000g for 60 min to separate cytoplasmic and membrane fractions. After 4 h of induction, 46% (11,340 U) of the above-described activity in the soluble fraction (24,653 U) was found in the cytoplasmic fraction, while 54% (13,312 U) was detected in the membrane fraction. After 8 h of induction, 24% of the soluble fraction (4617 U) was found in the cytoplasmic fraction, whereas 76% (14,622 U) was present in the membrane fraction. When we examined these activities after 20 h of induction, only 8% (850 U) activity was found in the cytoplasmic fraction, whereas 92% (9770 U) of the activity of the soluble fraction was detected in the membrane fraction. These results further validated that extracellular α-amylase activity was not the result of the cell lysis but translocation of mature protein through the outer membrane of the cell after cleavage by signal peptidase.
Archaeal enzymes have high demand in industry because of their tolerance to wide range of parameters including temperature, pH, and salinity [18]. However, it is arduous to get these enzymes from archaea on large scale because of poor growth and difficulties involved in growing these microorganisms. This is being circumvented by production of recombinant proteins from these microorganisms using various expression systems. Among them, the E. coli expression system is the most popular because in this system the recombinant proteins are produced in high quantities, culturing of the cells is easy and economical, the cells proliferate quickly, and they are easy to transform. However, expression of foreign genes in E. coli sometimes results in improperly folded and inactive gene products due to the rapid accumulation of the recombinant proteins in the cytoplasm [8-10, 19]. This can be averted if the recombinant proteins are secreted outside the cell. There are some reports that E. coli signal peptidases cleave the signal sequence of heterologous proteins from gram-positive bacteria including α-amylase [14, 20], subtilisin [21], mannase [22, 23], xylanase [15], and chitinase [23]. To the best of our knowledge, there is no report on archaeal signal peptide cleavage by bacterial signal peptidases prior to this study. It seems possible that signal peptide of Tk1884 can be used for the secretion of other heterologous recombinant proteins using the E. coli expression system.
REFERENCES
1.Tuteja, R. (2005) Type I signal peptidase: an
overview, Arch. Biochem. Biophys., 441, 107-111.
2.Schatz, G., and Dobberstein, B. (1996) Common
principles of protein translocation across membranes, Science,
271, 1519-1526.
3.Bardy, S. L., Eichler, J., and Jarrell, K. F.
(2003) Archaeal signal peptides – a comparative survey at the
genome level, Protein Sci., 12, 1833-1843.
4.Von Heijne, G. (1990) Protein targeting signals,
Curr. Opin. Cell Biol., 2, 604-608.
5.Brockmeier, U., Caspers, M., Freudl, R., Jockwer,
A., Noll, T., and Eggert, T. (2006) Systematic screening of all signal
peptides from Bacillus subtilis: a powerful strategy in
optimizing heterologous protein secretion in Gram-positive bacteria,
J. Mol. Biol., 362, 393-402.
6.Ng, S. Y., Chaban, B., VanDyke, D. J., and Jarrell,
K. F. (2007) Archaeal signal peptidases, Microbiology,
153, 305-314.
7.Lee, S. Y. (1996) High cell density culture of
Escherichia coli, Trends Biotechnol., 14,
98-105.
8.Marston, F. A. (1986) The purification of
eukaryotic polypeptides synthesized in Escherichia coli,
Biochem. J., 240, 1-12.
9.Thomas, J. G., and Baneyx, F. (1997) Divergent
effects of chaperone overexpression and ethanol supplementation on
inclusion body formation in recombinant Escherichia coli,
Protein Expres. Purif., 11, 289-296.
10.Hockney, R. C. (1994) Recent developments in
heterologous protein production in Escherichia coli, Trends
Biotechnol., 12, 456-463.
11.Makrides, S. C. (1996) Strategies for achieving
high-level expression of genes in Escherichia coli,
Microbiol. Rev., 60, 512-538.
12.Shokri, A., Sanden, A., and Larsson, G. (2003)
Cell and process design for targeting of recombinant protein into the
culture medium of Escherichia coli, Appl. Microbiol.
Biotechnol., 60, 654-664.
13.Choi, J. H., and Lee, S. Y. (2004) Secretory and
extracellular production of recombinant proteins using Escherichia
coli, Appl. Microbiol. Biotechnol., 64, 625-635.
14.Malik, B., Rashid, N., Ahmad, N., and Akhtar, M.
(2013) Escherichia coli signal peptidase recognizes and cleaves
the signal sequence of α-amylase originating from Bacillus
licheniformis, Biochemistry (Moscow), 78,
958-962.
15.Jalal, A., Rashid, N., Ahmed, N., Iftikhar, S.,
and Akhtar, M. (2011) Escherichia coli signal peptidase
recognizes and cleaves the signal sequence of xylanase from a newly
isolated Bacillus subtilis strain R5, Biochemistry
(Moscow), 76, 347-349.
16.Morikawa, M., Izawa, Y., Rashid, N., Hoaki, T.,
and Imanaka, T. (1994) Purification and characterization of a
thermostable thiol protease from a newly isolated hyperthermophilic
Pyrococcus sp., Appl. Environ. Microbiol., 60,
4559-4566.
17.Atomi, H., Fukui, T., Kanai, T., Morikawa, M.,
and Imanaka, T. (2004) Description of Thermococcus kodakaraensis
sp. nov., a well studied hyperthermophilic archaeon previously reported
as Pyrococcus sp. KOD1, Archaea, 1, 263-267.
18.Niehaus, F., Bertoldo, C., Kahler, M., and
Antranikian, G. (1999) Extremophiles as a source of novel enzymes for
industrial application, Appl. Microbiol. Biotechnol., 51,
711-729.
19.Rosano, G. L., and Ceccarelli, E. A. (2014)
Recombinant protein expression in Escherichia coli: advances and
challenges, Front. Microbiol., 5, 172.
20.Shahhoseini, M., Ziaee, A. A., and Ghaemi, N.
(2003) Expression and secretion of an α‐amylase gene from
a native strain of Bacillus licheniformis in Escherichia
coli by T7 promoter and putative signal peptide of the gene, J.
Appl. Microbiol., 95, 1250-1254.
21.Ikemura, H., Takagi, H., and Inouye, M. (1987)
Requirement of pro-sequence for the production of active subtilisin E
in Escherichia coli, J. Biol. Chem., 262,
7859-7864.
22.Zhang, Q., Yan, X., Zhang, L., and Tang, W.
(2006) Cloning, sequence analysis, and heterologous expression of a
β-mannanase gene from Bacillus subtilis Z-2, Mol.
Biol., 40, 368-374.
23.Yamabhai, M., Emrat, S., Sukasem, S., Pesatcha,
P., Jaruseranee, N., and Buranabanyat, B. (2008) Secretion of
recombinant Bacillus hydrolytic enzymes using Escherichia
coli expression systems, J. Biotechnol., 133,
50-57.
Supplementary Table and Figures (PDF)