Focus on Molecular Functions of Anti-Aging Deacetylase SIRT3
Jarmila Nahálková*
Biochemistry, Molecular, and Cell Biology Unit, Biochemworld Co., 74394 Skyttorp, Uppsala County, Sweden
Received November 11, 2021; Revised December 20, 2021; Accepted December 22, 2021
SIRT3 is a protein lysine deacetylase with a prominent role in the maintenance of mitochondrial integrity, which is a vulnerable target in many diseases. Intriguingly, cellular aging is reversible just by SIRT3 overexpression, which raises many questions about the role of SIRT3 in the molecular anti-aging mechanisms. Therefore, functions of SIRT3 were analyzed through the interaction network of 407 substrates collected by data mining. Results of the pathway enrichment and gene function prediction confirmed functions in the primary metabolism and mitochondrial ATP production. However, it also suggested involvement in thermogenesis, brain-related neurodegenerative diseases Alzheimer’s (AD), Parkinson’s, Huntington’s disease (HD), and non-alcoholic fatty liver disease. The protein node prioritization analysis identified subunits of the complex I of the mitochondrial respiratory chain (MRC) as the nodes with the main regulatory effect within the entire interaction network. Additional high-ranked nodes were succinate dehydrogenase subunit B (SDHB), complex II, and ATP5F1, complex V of MRC. The analysis supports existence of the NADH/NAD+ driven regulatory feedback loop between SIRT3, complex I (MRC), and acetyl-CoA synthetases, and existence of the nuclear substrates of SIRT3. Unexplored functions of SIRT3 substrates such as LMNA and LMNB; HIF-1a, p53, DNA-PK, and PARK7 are highlighted for further scientific advances. SIRT3 acts as a repressor of BACE1 through the SIRT3-LKB1-AMPK-CREB-PGC1A-PPARG-BACE1 (SIRT3-BACE1), which functions are fitted the best by the Circadian Clock pathway. It forms a new working hypothesis as the therapeutical target for AD treatment. Other important pathways linked to SIRT3 activity are highlighted for therapeutical interventions.
KEY WORDS: SIRT3, NAD+-dependent protein deacetylase, protein interaction network, pathway enrichment analysis, aging, respiratory electron transport chain, mitochondria, age-related diseaseDOI: 10.1134/S0006297922010035
Abbreviations: AD, Alzheimer’s disease; BACE1, beta-site amyloid-beta A4 precursor protein-cleaving enzyme 1; CC, Circadian Clock; HD, Huntington’s disease; KO, knockout; LMNA, lamin A; MRC, mitochondrial respiratory chain; NAFLD, non-alcoholic fatty liver disease; OXPHOS, oxidative phosphorylation; PD, Parkinson’s disease; PDH, pyruvate dehydrogenase; ROS, reactive oxygen species; TCA, tricarboxylic acid; wt, wild type.
INTRODUCTION
Reversion of the aging process or at least its delaying is a very ambitious task for science of many generations. From the public health point of view, not the extension of total length of the human life, but the improvement of health and vitality of aged people would be a desirable outcome of the research.
The sirtuin family is known for the significant functional link to aging and age-associated diseases with outstanding features exhibited by SIRT3, a NAD+-dependent protein lysine deacetylase with major role in the protection of mitochondrial integrity. The experiments using SIRT3 knockout (KO) mice confirmed hyperacetylation of most mitochondrial proteins, thus proving that SIRT3 is a major mitochondrial protein deacetylase [1]. The SIRT3 KO mice demonstrate potential power of SIRT3 in the protection against age-related diseases since the animals develop a whole range of diseases including cancer, neurodegenerative, cardiovascular, and metabolic diseases [2]. Mice display damaged mitochondrial energetics and reduced ATP production accompanied by the increased acetylation of mitochondrial proteins including MnSOD, which loses its capability to protect against increased levels of reactive oxygen species (ROS). The process leads ultimately to carcinogenesis, thus SIRT3 acts as a tumor suppressor since its deletion is sufficient to trigger tumorigenesis [3]. SIRT3 crucially regulates mitochondrial bioenergetics, which is activated by nutrients and exercise [4]. Its expression level declines in aging human stem cells (HSC-s) [5], frontal lobe, and hippocampus of the old rats [6]. Intriguingly, aging is reversible in the HSC-s just by SIRT3 overexpression, which provides evidence of its biological functions in the cellular anti-aging mechanisms [5]. SIRT3 also acts as a neuroprotective agent against the mitochondrial disruption by quenching ROS, preventing mitochondrial membrane potential loss, and as a sensor of neurotoxic insults in the rat models of Alzheimer’s disease (AD) and in the AD brain tissues [7].
To clarify molecular mechanism of the anti-aging effect of SIRT3, the present study explores the substrate interaction network of SIRT3. The analysis utilizes the hypothesis that the direct protein–protein interactions predict involvement in the same diseases and the interacting proteins are with high confidence participating in identical cellular and molecular functions [8]. The aim was achieved by the pathway enrichment analysis performed by the GeneMania application operating under the Cytoscape environment and complemented with the web-based STRING analysis. The protein node prioritization and the cluster analysis using Cyto-Hubba and MCODE applications identified high-priority protein nodes and clusters with major regulatory roles. The results of the analysis are discussed in the framework of anti-aging functions of the enzyme and its mitochondrial functions in the age-related diseases. The SIRT3 activity as a repressor of BACE1 (beta-site amyloid-beta A4 precursor protein-cleaving enzyme 1) through the SIRT3-LKB1-AMPK-CREB-PGC1A-PPARG-BACE1 (SIRT3-BACE1) pathway formed a new working hypothesis for the BACE1 regulation, one of the main therapeutical targets for AD treatment. The most important signaling and metabolic pathways linked to SIRT3 activity are highlighted for further regulatory options and therapeutical interventions.
MATERIALS AND METHODS
The list of SIRT3 substrates. SIRT3 substrates were collected from the literature sources [4, 9-29], which also included large-scale proteomic studies [30-32] (Tables S1 and S2 in the Supplement). One of the studies identified the differentially acetylated liver mitochondrial proteins isolated from the wild type (wt) and SIRT3 KO mice using LC/MS-MS analysis. The SIRT3 substrates with the two-fold increased acetylation level in the SIRT3 KO mice compared to the wt mice and p < 0.01 were selected for the analysis [30]. The further proteomic study used nano reverse-phase LC/MS-MS for identification of the SIRT3 substrates from the liver mitochondrial acetylome of the wt and SIRT3 KO mice. The proteins with minimum 2-fold acetylation increase in the SIRT3 KO compared to the wt mice and Welch’s t-test with Storey Correction <0.01 were chosen for further analysis [31]. Finally, the substrates from the SILAC studies of the differential protein acetylation between the SIRT3 KO and the wt murine embryonic fibroblasts and U2OS cells with SIRT3 upregulated by retroviral overexpression compared to the cells with shRNA silenced SIRT3 were included. The differential threshold was set to the 2-fold acetylation change for the ratios SIRT3KO/wt and SIRT3KO/SIRT3 overexpression [32]. The final list of SIRT3 substrates used for the analysis contained 407 proteins (Table S2 in the Supplement).
GeneMania analysis. The protein names of the SIRT3 substrates were converted to the GeneMania compatible symbols using the Protein Knowledgebase UniprotKB [33] and the database OMIM® (Online Mendelian Inheritance in Man®) [34]. GeneMania (3.5.2) [35-37] analysis was performed under Cytoscape (3.7.2) [38] environment utilizing the converted SIRT3 substrate list, which included SIRT3 as a query (Table S2 in the Supplement). The analysis was run against the H. sapiens database (update 13-07-2017) and by including all types of interaction networks. The analysis was set up for identification of the top 20 related genes and at the most 20 attributes by employing GO Molecular function weighting. The weighting method was selected based on high probability of sharing of the molecular functions between the SIRT3 substrates since the network of 408 proteins (including SIRT3) is interconnected by 1230 protein–protein interactions [8]. The category of results “Consolidated pathways” represents the result of the pathway enrichment, which is further used for the discussion.
String analysis. The web-based STRING analysis (v. 11) [39] was executed using the SIRT3 substrate list (Table S2) as a multiple protein query against the H. sapiens database. The settings were selected as follows: active interaction sources – text mining, experiments, and databases; minimum required interaction score – highest confidence (0.9); maximum number of interactors to show 1st and 2nd shell – none. The results of the functional enrichment analysis were exported with a False Discovery Rate (FDR) value less or equal to 0.05. The interaction network was clustered by the Markov Cluster Algorithm (MCL) clustering method [40] with the inflation value equal to 10.
The protein nodes prioritization. The high-priority essential protein nodes and pathways of the SIRT3 substrate interaction network constructed by GeneMania were predicted by the Cyto-Hubba application (0.1) [41]. The application was set up for the use of the topological method Maximum click centrality (MCC), which provides, according to the developers, the highest precision in the prediction of the high priority nodes [41]. Further, the settings of the analysis were adjusted for selection of the first-stage nodes and display of the shortest path. The first-stage nodes represent the protein nodes interacting directly with the query proteins.
Cluster analysis. Highly interconnected protein node clusters, which with high probability represent functional complexes were identified by the Cytoscape application MCODE (v. 1.6) [42]. The following software settings were used for the analysis: Network scoring – Degree cutoff: 2; Cluster finding – Haircut; Node score cutoff: 0.2; K-Core: 2; Max. depth: 100.
The pathway visualization. The pathway illustration was created by utilizing the knowledgebase of the WikiPathways application [43] run under Cytoscape (3.9.0) [38] environment and by using literature references as mentioned in the text. The drawing was performed by Adobe Illustrator 2020 (24.2.1).
RESULTS AND DISCUSSION
The present study explores a putative anti-aging function of the main mitochondrial protein deacetylase SIRT3 for potential therapeutical interventions. The main aim is to develop a working hypothesis by the processing large-scale data, which would be applicable for further experimental aging-related research.
The attractive rejuvenation ability of SIRT3 is apparent in the aged HSC-s, where it assists in the cell survival under oxidative stress and restoring youthful phenotype [5]. SIRT3 expression level in the young cells is high, however, it decreases with aging of the cells and tissues, which leads to the increased ROS levels and oxidative damages [6, 44]. SIRT3 KO mice display an overwhelming range of age-related diseases [2] and due to the mentioned anti-aging effect of SIRT3 [5], pharmacological activation of SIRT3 and regulation of the functionally linked pathways are highly demanded for therapeutical advancements.
To investigate the mentioned anti-aging effects, this study explores the protein interaction network of SIRT3 substrates using pathway enrichment, protein function prediction, protein node prioritization, and clustering methods (Fig. 1, Fig. 2, Table 1). The interaction network analysis facilitates discovery of the most significant molecular functions associated with SIRT3 deacetylation activity. The use of artificial intelligence is especially helpful for retrieving information from the limited literature sources, which could remain unnoticed by the conventional data mining. New hypotheses and conclusions can be created based on the obtained data, which are suitable for further experimental validation.
Fig. 1. Pathway enrichment analysis of the SIRT3 substrate interaction network performed by the web-based STRING application. a) KEGG Pathways; b) GO Cellular Component. The analysis was performed using the SIRT3 substrate list as a multiple protein query against the H. sapiens database.
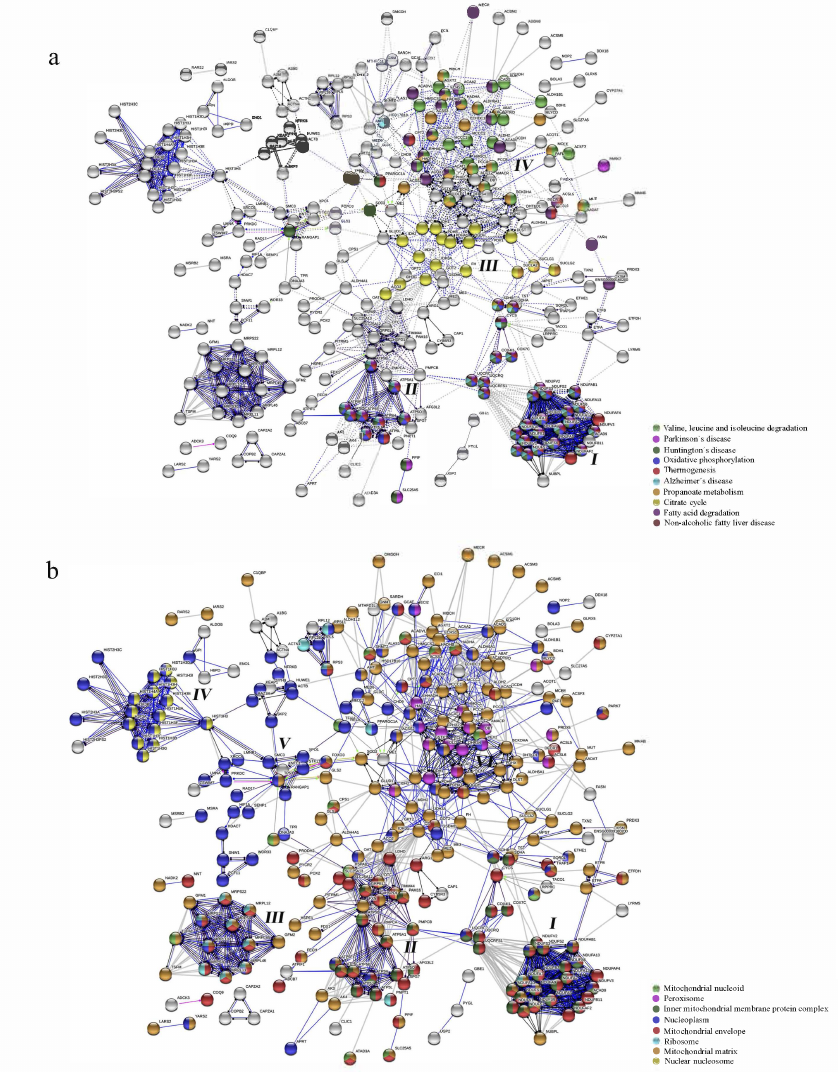
Fig. 2. Protein node prioritization of the SIRT3 substrate interaction network predicted by the CytoHubba (0.1) application. The network was constructed by applying the GeneMania (3.5.2) application run under Cytoscape (3.7.2) environment utilizing the H. sapiens database (update July 13, 2017) and including physical interactions only. The proteins are shown from the highest (red) to the lowest (yellow) ranked nodes.
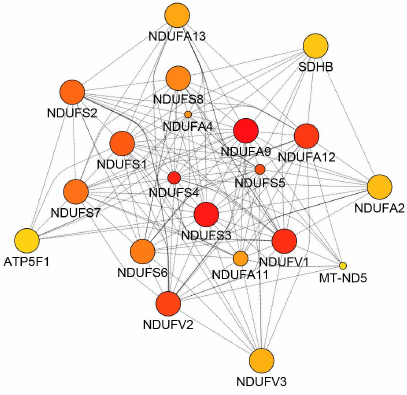
Table 1. Results of the pathway enrichment
analysis and the gene function prediction analysis of the SIRT3
substrate interaction network performed by the GeneMania application
run under the Cytoscape (3.7.2) environment
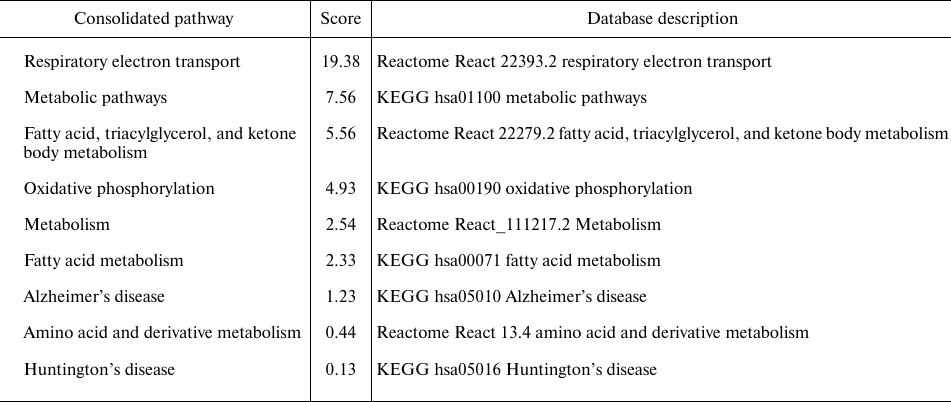
Notes. The analysis was performed by utilizing the H. sapiens
database (update July 13, 2017) and by including all types of
interaction networks.
* Score (weight) expresses the predictive value, which GeneMania assigns
to the pathways, how well they correspond to the query dataset compared
to the non-query.
The protein node prioritization is employed to determine the high-priority protein nodes and clusters of the interaction network with major regulatory roles. Attention is also paid to the subcellular localization of the substrates processed by SIRT3, which assists in the additional interpretation of SIRT3 functionality in different cellular compartments. Finally, a working model of a pathway for suppression of BACE1 activity by SIRT3 was constructed for further developments of AD therapies.
SIRT3 substrate interaction network. The present analysis utilizes Cytoscape application GeneMania, pathway enrichment, and gene function prediction analytical tool, which is based on the combined database annotations of genomic, proteomic, and mRNA expression data [35, 36, 45].
The software matches the query substrate list with the predicted and experimentally confirmed physical protein–protein interactions, genetic interactions, results of co-localization, and co-expression studies. Finally, it fits the network with the known signaling and metabolic pathways. One of the advantages of the GeneMania application is its integration with the Cytoscape environment, which allows efficient construction of the interaction networks. The GeneMania database is continuously updated and expanded with the additional data [37], which is not always the case with other types of interaction network platforms.
The outcome of the GeneMania analysis matched the most important functions of the SIRT3 substrate interaction network, which are linked to the respiratory electron transport chain, general metabolic pathways, fatty acid, triacylglycerol, and ketone body metabolism, whereas AD and HD obtained lower scores (Table 1). The network was further subjected to the protein node prioritization by the CytoHubba application. The subunits of NADH:ubiquinone oxidoreductase, representing complex I of the mitochondrial respiratory chain (MRC) were identified among the nodes with the highest rank (Fig. 2). It suggests that they have the most important regulatory effect within the entire SIRT3 substrate interaction network (Table 1). Additional high-ranked nodes are succinate dehydrogenase subunit B (SDHB), complex II, and ATP5F1, which encodes the subunit B of the ATP synthase Fo unit, complex V of the MRC. MCODE cluster analysis identified among the most important interaction subgroups the priority subnetwork of 70 nodes involved in the oxidative phosphorylation (OXPHOS), respiratory transport chain, and the interaction networks of the diseases AD and HD (data not shown), which defines the main regulatory nodes of the interaction network.
The GeneMania analysis was further complemented with the results of the pathway enrichment using the web-based STRING database, which yielded a highly interconnected protein–protein interaction network of the SIRT3 substrates (Fig. 1). The algorithm complements the protein dataset of SIRT3 substrates with known interactions from the public databases and through computational predictions. The system further performs classification based on the Gene Ontology, KEGG, high-throughput text mining, and hierarchical clustering of the network [39, 46, 47], which provides useful outcomes for many biological questions.
Additional results of the STRING analysis using the KEGG pathways classification visualized a linkage of the SIRT3 substrates to Parkinson’s disease (PD), Huntington’s disease (HD), Alzheimer’s disease (AD), and non-alcoholic fatty liver disease (NAFLD), while creating discrete clusters by the MCL method (Fig. 1a) [40, 48]. High expression of SIRT3 in the metabolically active tissues such as brain, liver, heart, and brown adipose tissues (BAT) correlates with the functional roles in the neurodegenerative pathologies, NAFLD, and the process of thermogenesis (Fig. 1a) [1].
The interaction network analyzed based on the KEGG Pathways classification and clustered by the MCL method (Fig. 1a) was split into four clusters, which were correlated with the subcellular localization of SIRT3 substrates (five clusters, Fig. 1b). Cluster I contains the SIRT3 substrates associated with OXPHOS, AD, PD, HD, and NAFLD (Fig. 1a). It consists of NADH:ubiquinone oxidoreductase (complex I) subunits, an enzyme that performs electron transfer from NADH to ubiquinone acceptor, part of the respiratory electron transfer chain (Fig. 1a). The complex I activity is inhibited in the SIRT3 KO mice, while the external SIRT3 can restore it, which suggests activation effect occurring certainly through the deacetylase activity of SIRT3. The ATP production is affected more than 50% in these mice and ~30% in the isolated MEF (mouse embryonic fibroblast) cells [49]. This result could be related to the present study, which shows that SIRT3 deacetylates multiple subunits of the complex I, II, and even of the complex V of MRC (Cluster I and II, Fig. 1a), which overlap with the priority substrates of the network constructed by GeneMania (Fig. 2). However, it seems that SIRT3 regulates the cellular mitochondrial energetics by its deacetylase activity only partially since its KO does not reduce the cellular ATP level completely. The present analysis supports existence of the NADH/NAD+ driven regulatory feedback loop between SIRT3, complex I (MRC), and acetyl-CoA synthetases [49]. SIRT3 requires for its activity NAD+ generated by the complex I MRC, which, in turn, is activated by SIRT3 deacetylase activity and creates the regulatory feedback loop [49]. Remarkably, SIRT3 and other sirtuins also activate by deacetylation a range of Acetyl-CoA synthetases [10], (Table S2 in the Supplement, Columns 24-32), which are acetyl donors for the protein acetylation including complex I, II, and V, MRC, and suppliers of acetyl-CoA. With regards to this function, SIRT3 is superior to other sirtuins in deacetylation of ACSS2 [10] and creates additional tuning of the mitochondrial energy production by controlling the acetyl-CoA synthesis. One of the ACSS2 substrates processes acetate and fatty acids into acetyl-CoA mostly used for oxidation in the tricarboxylic acid (TCA, Krebs) cycle and by this way significantly contributes to NADH and ATP production [50]. NADH is further used by the MRC complex I to produce proton gradient for ATP synthase and NAD+, which completes the regulatory loop. Prioritization analysis of the interaction network confirmed the highest significance of these mechanisms linked to the SIRT3 deacetylase activity with the highest impact on cellular energetics.
The cluster also involves the subunits of succinate dehydrogenase complex [subunit A (SDHA), and subunit B (SDHB)], MRC complex II. The result of the present analysis supports the results of processing of the complex II subunits by SIRT3 suggested by some authors [51]. Physical interaction of SIRT3 and SDHB was also demonstrated by another two independent experiments, however without detection of the specific acetylation sites [52]. Another report showed that deacetylation of SDHA by SIRT3 has only a partial stimulation effect on the activity of the whole complex II [53] or it was not detected [49]. The mentioned limited regulation by acetylation makes sense since MRC complex II is an essential enzymatic complex and its complete inactivation is lethal [53].
Cluster II contains the substrates linked to AD, PD, and HD, which overlap with the functions in thermogenesis and OXPHOS (Fig. 1a). The cluster consists of the subunits of ATP synthase (complex V), the last enzyme of the mitochondrial OXPHOS chain, which produces cell energy in the form of ATP by chemiosmotic proton efflux. The increased acetylation of the enzyme subunits occurs in the mitochondria of the SIRT3 KO mouse livers and muscles, while the animals exhibit deficiency of mitochondrial ATP production [4]. It indicates that SIRT3 deacetylates and crucially regulates activity of the MRC complex V. On the other hand, SIRT3 stimulated by nutrients, calorie restriction (CR), and exercise deacetylates ATP-synthase subunits a, b, c, d, and OSCP, which leads to the increased mitochondrial energy supply and anti-aging effects [4]. Strikingly, SIRT3 is essential for the maintenance of the membrane potential of the healthy mitochondria, where it is bound to ATP synthase by pH and stress-sensitive bond [54]. Binding between SIRT3 and ATP5O can be disrupted by the change of the inner mitochondrial membrane potential, but not by inhibition of ATP production activity of the ATP synthase. Instead, ATP5O is one of the SIRT3 substrates (Table S2 in the Supplement), and as with other ATP synthase subunits (Table S2 in the Supplement, Lines 59-68), its activity is regulated through deacetylation by SIRT3, which suggests that ATP5O-SIRT3 binding is mediated by the enzyme–substrate interactions with SIRT3. The mitochondrial membrane potential is SIRT3 dependent, since after its disruption it recovers quickly in the wt HeLa cells, but not in the SIRT3 KO cells [54]. The ATP5F1 subunit is one of the top priority nodes of the SIRT3 substrate interaction network (Fig. 2), which together with the role in deacetylation of other subunits of the MRC complex V demonstrates the mentioned essential role of SIRT3 in mitochondrial bioenergetics.
Cluster III consists of the enzymes involved in TCA cycle not connected by STRING to any of the major network-related diseases (Fig. 1a), however, they have high importance in both cancer and AD. Glucose metabolism is affected significantly in the AD brains, where the most downregulated enzymes of the TCA cycle are located upstream of the succinyl-CoA [55]. PDHX, PDHB, PDHA1 (Fig. 1a, cluster III) are the subunits of the pyruvate dehydrogenase enzyme complex (PDHC), which is implicated in the first and irreversible catalytic step between the glycolysis and TCA cycle converting pyruvate to the acetyl-CoA. SIRT3 crucially activates the PDHA1 enzyme subunit by its deacetylation, which has importance for regulation of the Warburg effect in the cancer cells [56, 57]. The Aβ deposits of the AD brain cause increased phosphorylation of PDHC leading to the reduced energy outcome due to the metabolic switch from OXPHOS to glycolysis. Due to the restricted metabolic pathway, the alternative energy is obtained through the reversal enzymatic reaction converting oxaloacetate into succinate and by the electron transport chain of the complex I [58]. The age-related stimulation of pyruvate dehydrogenase kinase (PDK) deactivates PDHC activity through its phosphorylation and the resulting ATP production deficits cause mitochondrial damage and synaptic deterioration [59]. Since the expression of SIRT3 tends to decrease in aging cells and tissues [5, 6], aging is associated with the reduced mitochondrial ATP production due to the decreased activity of OXPHOS.
Cluster IV is formed by the SIRT3 substrates involved in fatty acid degradation and branched-chain amino acid metabolism (Fig. 1a). The increased acetylation of PDH in the SIRT3 KO mice leads to the metabolic switch through the increased fatty acid oxidation and accumulation of the lactate occurring in muscles [24]. It also causes hyperacetylation of the long-chain Acyl-CoA dehydrogenase, which leads to accumulation of the long-chain fatty acid metabolic intermediates [20]. It suggests that SIRT3 deacetylation activity has a broader regulatory effect including not only glucose metabolism, but also metabolism of fatty acids and branched amino acids.
Subcellular localization of SIRT3 and its substrates. Mostly described as a mitochondrial protein deacetylase, there is a disagreement on subcellular localization of SIRT3 in other compartments, which is a debated topic. According to some reports, SIRT3 shuttles between the nucleus and mitochondria, which occurs upon exposure to a cellular stressor or after overexpression. Before it is exported to mitochondria, SIRT3 is processed by the N-terminal cleavage of 142 amino acid residues [60]. Nuclear localization of SIRT3 was, however, previously doubted by some authors since the deacetylase is exclusively mitochondrial according to their experimental data [61, 62].
To evaluate subcellular localization of SIRT3 activity, the STRING interaction network of the collected SIRT3 substrates was processed through the GO Cellular Component classification. The analysis showed four main subcellular compartments as locations of the SIRT3 substrates, which were nucleus, mitochondria, cytoplasm, and peroxisomes (Fig. 1b). Majority of the SIRT3 substrates also co-localized in the cytoplasm (data not shown) excluding histone proteins. The SIRT3 substrates used for data mining originated from the analysis of the mitochondrial fractions [30, 31], where the mitochondrial proteins are overrepresented. The exception is the SILAC study [32] (Table S1), which used the whole-cell lysis step before the LC-MS/MS identification. The SIRT3 substrate list obtained from this study contains multiple histone proteins (Fig. 1b), while their occurrence in the lists of the substrates from other sources [30, 31] is limited.
Interestingly, clusters I and II (Fig. 1b) contain both mitochondrial and nuclear SIRT3 substrates. The complex I respiratory electron transport chain subunits (NDUFS1-3, NDUFS6), acyl-CoA dehydrogenase subunit 9 (ACAD9), and NDUFAB1 are located both in mitochondria and nucleoplasm. The ATP synthase peripheral stalk membrane subunit B (ATP5F1) is also localized in nucleoplasm, mitochondrial membrane, and mitochondrial matrix (Fig. 1b). PNPT1 is involved in the processing of the mitochondrial mRNA and polyadenylation occurs in the mitochondrial ribosomes and mitochondrial envelope [63]. SLC25A13, a mitochondrial calcium-binding carrier, transports the cytoplasmic glutamate for the mitochondrial aspartate through the inner mitochondrial membrane [64], located in the mitochondrial matrix and mitochondrial nucleoid (Fig. 1b).
Cluster III contains mitochondrial proteins encoded in the nucleus and localized in the different mitochondrial compartments (Fig. 1b). SIRT3 substrates GFM1, GFM2, and TSFM included in these clusters are the mitochondrial elongation factors important for the protein translation in the mitochondria and they are localized in the mitochondrial matrix. Their mutations are causative agents of mitochondrial diseases due to MRC deficiencies [65-67]. Another translation elongation factor, TUFM [68], is localized both in the matrix and nucleoid of the mitochondria (Fig. 1b). SIRT3 was demonstrated to regulate translational activity of the mitochondrial ribosomes [19]. The mitochondrial ribosomal proteins MRPL10, MRPL11, MRPL12, MRPL45, MRPL46, MRPS22, MRPS26, MRPS30, and MRPS9 involved in this cluster are encoded by the nuclear DNA.
Cluster IV contains nuclear histone proteins and several exclusively nucleoplasmic proteins (Fig. 1b). The enzymatically active SIRT3 fulfills the function in the chromatin silencing by deacetylation of H4K16Ac, H3K9Ac [60], and H3K56Ac [69], which also supports existence of the functional SIRT3 in the nucleus. Interestingly, co-expression with SIRT5 relocates SIRT3 from mitochondria to the nucleus [70]. Among the sirtuins, SIRT3 has the best ability to remove β-hydroxybutyryl (bhb) from the nuclear substrate H3K9bhb [63] and it also deacetylates the nuclear substrate H3K56 [64].
Cluster V includes several remarkable SIRT3 substrates with interesting molecular functions located in the nucleoplasm such as LMNA (lamin A), LMNB, and HIF-1A, p53, and PRKDC, which are highlighted further. The SIRT3 substrate LMNA was identified by the SILAC method in the U2OS cells overexpressing SIRT3 versus the KO cells, while LMNB was found by the identical technique in the SIRT3 KO MEF compared to the wt cells (Table S1 in the Supplement) [32]. Interestingly, similar to the aging-related functions of SIRT3, mutations of its substrate, LMNA, cause the syndrome of premature aging called Hutchinson–Gilford progeria syndrome [71]. Moreover, LMNB1 K134 acetylation controls nuclear stability and cell cycle progression. It induces persistent activation of the G1/S DNA damage checkpoint, cell cycle arrest at G1, negatively regulates aberrant canonical non-homologous end joining (cNHEJ), and causes increased association of LMNB1 with the nuclear periphery proteins [72]. The cells depleted in LMNA/C also show aberrations in the DNA base excision repair (BER) [73], DNA double-strand break (DSB) repair, and homologous recombination (HR) [71]. Due to the range of interesting functions of lamins and lack of knowledge about their SIRT3 deacetylation, related research should be encouraged.
An additional interesting substrate included in this cluster is HIF-1a (Table S1 in the Supplement) [32], which is also deacetylated by SIRT1 at K674 [74]. SIRT3 is a crucial regulator of cancer cell metabolism since it mediates metabolic switch towards glycolysis by destabilization of HIF-1a through its deacetylation. The knockout increases ROS levels, stabilizes HIF-1a, and increases the Warburg effect, while overexpression acts as a tumor suppressor by inhibiting glycolysis and cancer cell proliferation [13, 75].
Another significant substrate involved in this cluster is p53, a classical tumor suppressor, which was already identified as a SIRT3 substrate in the PTEN deficient cancer cells [76]. Amazingly, the whole biological process of neurodegeneration can be reversed just by the increase of SIRT3 expression, since it prevents damage of the neuronal mitochondria caused by co-expression with p53 [77]. The SIRT3 activation can effectively decrease the neurodegenerative damages caused by the upregulation of p53, which is promising for future research.
Additional substrate, the DNA-dependent protein kinase (PRKDC, DNA-PK) [30, 32], is a nuclear enzyme involved in the end-joining of double-strand DNA break repair (NHEJ) [78, 79]. The DNA-PK is also involved in the maintenance of the telomere length and their protection, which has great importance both in cancer and aging. During aging, when DNA breaks accumulate, DNA-PK activation leads to deterioration of the mitochondrial bioenergetics and muscle fitness [80, 81]. The common feature with SIRT3 is the effect of calorie-restriction diet and aerobic exercise, which can decrease the DNA-PK activity during aging [80]. On the other hand, SIRT3 is stimulated by nutrients, CR, and exercise, which leads to the increased mitochondrial energy supply and anti-aging effects [4]. Functional consequences of the SIRT3-mediated deacetylation of DNA-PK on mitochondrial energy production and DNA repair mechanisms are not known, despite that, they could be partially predicted from their main functions. They could be an interesting subject for further research.
The PARK7 deacetylation could also be potentially interesting for further experimental examination. Mutations in the PARK7 gene are causative agents of the early onset of PD [82], which is one of the important diseases associated with the SIRT3 substrate interaction network (Fig. 1a).
Cluster VI is defined by molecular functions involved in inactivation of ROS and β-oxidation of fatty acids and unique peroxisome localization, which should be experimentally clarified. The substrates might firstly, however, be processed in mitochondria and nuclei before their relocation to peroxisomes. This observation is supported by the existence of membrane contact sites of peroxisome membranes with other organelles including mitochondria and nuclei [83].
In conclusion, the collected SIRT3 substrates were sorted into the distinct clusters based on their subcellular localization in the mitochondrial, nuclear, and peroxisomal subcellular compartments. In addition to the cluster of the solely nuclear histone substrates, other nuclear-encoded proteins with significant functions in the mitochondria are deacetylated by SIRT3. Present analysis supports the experimental observations that SIRT3 processes the substrates in the nucleus and not exclusively in the mitochondria. Several new nuclear substrates of SIRT3 with potentially interesting biological functions including LMNA, LMNB, HIF-1a, DNA-PK, and PARK7 are highlighted for further experimental studies. The peroxisomal membrane fusion with other organelles might be a feasible explanation of the occurrence of the SIRT3 substrates in peroxisomes, however, further experimental clarification will be required.
Regulation of BACE1. Related to the molecular function in AD pathology (Table 1), activation of the SIRT3 enzymatic activity remarkably inhibits Aβ production in the brain through inhibition of β-secretase (BACE1) [84], an enzyme of the first and the rate-limiting step in the amyloid precursor protein (APP) processing. The pathway facilitating the regulatory effect of SIRT3 on the activity of BACE1 was constructed using the knowledgebase WikiPathways, GeneMania application, and literature mining. This allowed creation of a working model for the mechanism of inhibition of the Aβ production in the brain (Fig. 3).
Fig. 3. Hypothetical mechanism of the inhibition of Aβ production in the brain. It shows regulation of β-secretase (BACE1) through the constructed SIRT3-LKB1-AMPK-CREB-PGC1A-PPARG-BACE1 pathway. The pathway illustration was created utilizing the knowledgebase of the application WikiPathways run under Cytoscape (3.7.2) environment and literature references [18, 84-87].
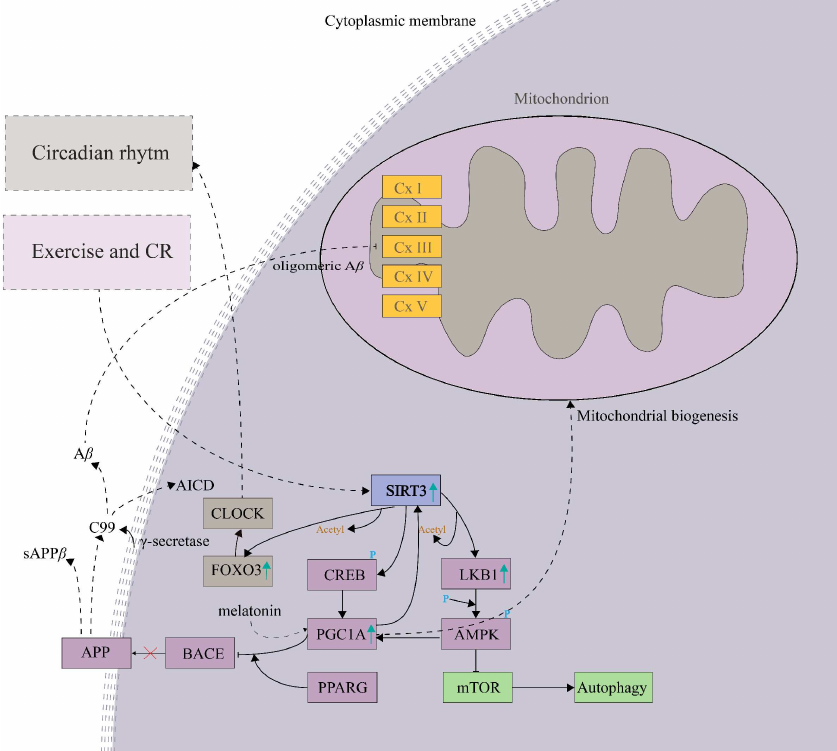
In first step, SIRT3 activated by exercise and/or CR deacetylates and activates the serine/threonine-protein kinase 11 (LKB1, STK11; Table S2 in the Supplement), which is followed by the AMPK phosphorylation and activation of the SIRT3-LKB1-AMPK pathway [18]. It results in upregulation of PGC-1α [84], a repressor of BACE1, which inhibits the production of Aβ in the brain [85] (Fig. 3). Alternatively, SIRT3 also activates CREB, which stimulates the PGC-1α promoter directly [86]. PGC-1α further requires the presence of PPARG for its effect on the expression of BACE1. In agreement with this model, the expression level of PGC-1α is decreased in the hippocampus of AD patients compared to the age-matched controls [87], which supports relevance of the hypothetical SIRT3 – LKB1 – AMPK – CREB – PGC1A – PPARG – BACE1 pathway (Fig. 3).
The molecular functions mediated by the SIRT3-LKB1-AMPK-CREB-PGC1A-PPARG-BACE1 pathway (Fig. 3) [18, 84-87] were analyzed by the pathway enrichment analysis performed employing GeneMania application. Interestingly, the highest score (weight) in the pathway enrichment was obtained by the Circadian Clock (CC) (Table 2). It might be due to the function of PGC-1α, which mediates the inhibitory effect on BACE1 activity. PGC-1α also plays a central role in the regulation of the key oscillators of circadian rhythm BMAL1 and CLOCK, it coordinates functions of the mammalian clock and energy metabolism [87].
Table 2. Results of the pathway enrichment
analysis performed by GeneMania application run under the Cytoscape
(3.7.2) environment, where all proteins of the
SIRT3-LKB1-AMPK-CREB-PGC1A-PPARG-BACE1 pathway were used as an
input
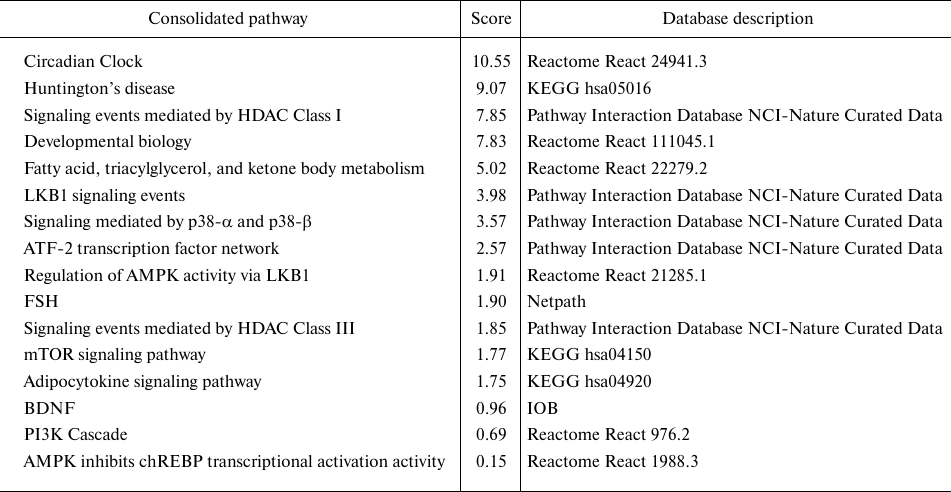
Notes. The analysis was run against the H. sapiens database
(update July 13, 2017) and by including all types of interaction
networks. The circadian clock pathway obtained the highest score in the
analysis.
* Score (weight) expresses the predictive value, which GeneMania assigns
to the pathways, how well they correspond to the query dataset compared
to the non-query.
Interestingly, the circadian rhythm regulator melatonin also activates PGC-1α, which further interacts with the estrogen-related receptor α (ERRα) directly bound on the SIRT3 promoter and enhances its transcription activity [88]. Furthermore, CC regulates the rhythm of NAD+-dependent SIRT3 through the nicotinamide riboside (NR) pathway, which is activated by nutrient availability during the feeding period [89]. The CC disruption can be a causative agent of the diseases associated with the SIRT3 substrate interaction network (Fig. 1a) AD, PD, and HD [90].
Interestingly, acetylation of the cellular proteins also exhibits diurnal patterns since most of the nuclear and cytoplasmic proteins are acetylated during the night, and mitochondrial proteins are modified during the day. The rhythmic acetylation is functionally linked to the rhythmic deacetylation, while SIRT3 is the main mitochondrial protein deacetylase [89]. Therefore, SIRT3 might function as a rhythmic circadian deacetylase of the nuclear and mitochondrial proteins with the potential effect on neurodegenerative disease development, which could occur through the nodes of the SIRT3-BACE1 pathway.
The CC regulation also occurs through the SIRT3-FOXO3-CLOCK pathway (Fig. 3), where FOXO3 is a crucial regulator. Under conditions of low insulin occurring in the aging brain [91], it binds directly to the CLOCK promoter and regulates its transcription [92]. The SIRT3 activation increases FOXO3 expression through their direct physical interaction, which, however, does not depend on the SIRT3 deacetylase activity [93, 94].
In a conclusion, function of the SIRT3-LKB1-AMPK-CREB-PGC1A-PPARG-BACE1 pathway is closely related to the CC pathway, which deteriorates in AD, PD, and HD and it appears to be one of the causative agents of the diseases. The SIRT3-FOXO3-CLOCK pathway associated with metabolic disruption, ROS production, and autophagy, is also relevant to the AD pathology. The pathways mediate mutual regulatory effect of protein nodes, which could be potentially attractive for multitarget therapy development.
CONCLUSIONS
The current interaction network analysis of the substrates of the main mitochondrial deacetylase SIRT3 highlighted AD, PD, HD, and NAFLD as the diseases most significantly associated with its deacetylation activity. The confirmed molecular functions associated with the SIRT3 substrates are within the respiratory electron transport chain, TCA cycle, fatty acid, triacylglycerol, and ketone body metabolism. SIRT3-mediated deacetylation occurs predominantly in BAT, where it contributes to adaptive thermogenesis, a biological process emphasized by the analysis.
SIRT3 deacetylates multiple subunits of the complex I, II, and even complex V of MRC, while the role of complex I deacetylation has the most important regulatory effect within the entire interaction network of the analyzed substrates. The analysis supports existence of the NADH/NAD+ driven regulatory feedback loop involving SIRT3, complex I (MRC), and acetyl-CoA synthetases. SIRT3 also crucially deacetylates and activates the key enzymes of the TCA cycle, such as PDHA1, responsible for the switch from OXPHOS to glycolysis. It can cause deterioration of energy production during aging when the expression of SIRT3 decreases and ultimately leads towards the decrease of the cellular energy level. Deacetylation of the subunits of the complex II and complex V finally contributes to the regulation of the mitochondrial ATP production. It seems that other factors also contribute to the regulation, since SIRT3 KO does not cause complete collapsing of the mitochondrial bioenergetics, which would be interesting to investigate further.
Interestingly, the analysis also revealed several understudied nuclear substrates with biological functions involved in the maintenance of the nuclear architecture and stability, genomic integrity, cell cycle progression, and DNA repair. Deacetylation of LMNA and LMNB (HGPS, Hutchinson–Gilford Progeria syndrome); HIF-1a (Warburg effect), p53 (neuroprotection), DNA-PK (NHEJ), and PARK7 (familiar PD) together with other substrates involved in the functions of mitochondrial bioenergetics could possibly contribute to the anti-aging activity of SIRT3.
In the brain tissues, SIRT3 performs as a repressor of BACE1, an enzyme that catalyzes crucial step of the Aβ peptide production, one of the hallmarks of AD. The constructed regulatory pathway SIRT3-LKB1-AMPK-CREB-PGC1A-PPARG-BACE1 creates a model of the inhibitory action of SIRT3 on the enzymatic activity of BACE1 and it offers several alternative nodes for multitargeting by pharmaceuticals. The pathway is closely related to CC function, which also deteriorates during AD advancement, and is a direct causative agent of pathological neurodegenerative changes. Additional functions related to AD and CC are also fulfilled by the SIRT3-FOXO3-CLOCK pathway.
SIRT3 exhibits several modes of neuroprotective actions in the brain and liver including prevention of mitochondrial damages due to the respiratory electron transfer chain failure, quenching of ROS, inhibition of the mitochondrial membrane potential loss, and regulation of mitophagy. The increased expression and activity of SIRT3 in the brain and liver seems particularly beneficial to counter-balance negative effects of aging. Control of the liver functions of the complex I and II of MRC is also beneficial for the management of NAFLD.
Pharmacological activation of SIRT3 in combination with the stimulating effect of regular exercise is an attractive option for the improvement of negative consequences of aging. Increase of the mitochondrial bioenergy production through the priority nodes of the SIRT3 substrate interaction network is also a promising strategy to achieve the antiaging effect.
Acknowledgments. The research was financially supported by the Biochemworld Co., Uppsala County, Sweden.
Ethics declarations. The author declares no conflicts of interest. This article does not contain description of studies with the involvement of humans or animal subjects performed by the author.
Supplementary information. The online version contains supplementary material available at https://doi.org/10.1134/S0006297922010035.
REFERENCES
1.Lombard, D. B., Alt, F. W., Cheng, H.-L.,
Bunkenborg, J., Streeper, R. S., et al. (2007) Mammalian Sir2 homolog
SIRT3 regulates global mitochondrial lysine acetylation, Mol. Cell.
Biol., 27, 8807-8814, doi: 10.1128/mcb.01636-07.
2.McDonnell, E., Peterson, B. S., Bomze, H. M., and
Hirschey, M. D. (2015) SIRT3 regulates progression and development of
diseases of aging, Trends Endocrinol. Metab., 26,
486-492, doi: 10.1016/j.tem.2015.06.001.
3.Zhu, Y., Yan, Y., Principe, D. R., Zou, X.,
Vassilopoulos, A., et al. (2014) SIRT3 and SIRT4 are mitochondrial
tumor suppressor proteins that connect mitochondrial metabolism and
carcinogenesis, Cancer Metab., 2, 15, doi:
10.1186/2049-3002-2-15.
4.Vassilopoulos, A., Pennington, J. D., Andresson,
T., Rees, D. M., Bosley, A. D., et al. (2013) SIRT3 deacetylates ATP
synthase F1 complex proteins in response to nutrient- and
exercise-induced stress, Antioxid. Redox Signal., 21,
551-564, doi: 10.1089/ars.2013.5420.
5.Brown, K., Xie, S., Qiu, X., Mohrin, M., Shin, J.,
et al. (2013) SIRT3 reverses aging-associated degeneration, Cell
Rep., 3, 319-327, doi: 10.1016/j.celrep.2013.01.005.
6.Braidy, N., Poljak, A., Grant, R., Jayasena, T.,
Mansour, H., et al. (2015) Differential expression of sirtuins in the
aging rat brain, Front Cell Neurosci., 9, 167, doi:
10.3389/fncel.2015.00167.
7.Weir, H. J. M., Murray, T. K., Kehoe, P. G., Love,
S., Verdin, E. M., et al. (2012) CNS SIRT3 expression is altered by
reactive oxygen species and in Alzheimer’s disease, PLoS
One, 7, 3-9, doi: 10.1371/journal.pone.0048225.
8.Oti, M. (2006) Predicting disease genes using
protein–protein interactions, J. Med. Genet., 43,
691-698, doi: 10.1136/jmg.2006.041376.
9.Poulose, N., and Raju, R. (2015) Sirtuin regulation
in aging and injury, Biochim. Biophys. Acta, 1852,
2442-2455, doi: 10.1016/j.bbadis.2015.08.017.
10.Hallows, W. C., Lee, S., and Denu, J. M. (2006)
Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases,
Proc. Natl. Acad. Sci. USA, 103, 10230-10235, doi:
10.1073/pnas.0604392103.
11.Shimazu, T., Hirschey, M. D., Hua, L.,
Dittenhafer-Reed, K. E., Schwer, B., et al. (2010) SIRT3 deacetylates
mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates
ketone body production, Cell Metab., 12, 654-661, doi:
10.1016/j.cmet.2010.11.003.
12.Bharathi, S. S., Zhang, Y., Mohsen, A. W.,
Uppala, R., Balasubramani, M., et al. (2013) Sirtuin 3 (SIRT3) protein
regulates long-chain acyl-CoA dehydrogenase by deacetylating conserved
lysines near the active site, J. Biol. Chem., 288,
33837-33847, doi: 10.1074/jbc.M113.510354.
13.Finley, L. W. S. S., Carracedo, A., Lee, J.,
Souza, A., Egia, A., et al. (2011) SIRT3 opposes reprogramming of
cancer cell metabolism through HIF1α destabilization, Cancer
Cell, 19, 416-428, doi: 10.1016/j.ccr.2011.02.014.
14.Sundaresan, N. R., Samant, S. A., Pillai, V. B.,
Rajamohan, S. B., and Gupta, M. P. (2008) SIRT3 is a stress-responsive
deacetylase in cardiomyocytes that protects cells from stress-mediated
cell death by deacetylation of Ku70, Mol. Cell. Biol.,
28, 6384-6401, doi: 10.1128/mcb.00426-08.
15.Qiu, X., Brown, K., Hirschey, M. D., Verdin, E.,
and Chen, D. (2010) Calorie restriction reduces oxidative stress by
SIRT3-mediated SOD2 activation, Cell Metab., 12, 662-667,
doi: 10.1016/j.cmet.2010.11.015.
16.Yu, W., Dittenhafer-Reed, K. E., and Denu, J. M.
(2012) SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and
regulates mitochondrial redox status, J. Biol. Chem.,
287, 14078-14086, doi: 10.1074/jbc.M112.355206.
17.Schlicker, C., Gertz, M., Papatheodorou, P.,
Kachholz, B., Becker, C. F. W., et al. (2008) Substrates and regulation
mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5, J.
Mol. Biol., 382, 790-801, doi:
10.1016/j.jmb.2008.07.048.
18.Pillai, V. B., Sundaresan, N. R., Kim, G., Gupta,
M. M. P., Rajamohan, S. B., et al. (2010) Exogenous NAD blocks cardiac
hypertrophic response via activation of the SIRT3-LKB1-AMP-activated
kinase pathway, J. Biol. Chem., 285, 3133-3144, doi:
10.1074/jbc.M109.077271.
19.Yang, Y., Cimen, H., Han, M. J., Shi, T., Deng,
J. H., et al. (2010) NAD+-dependent deacetylase SIRT3
regulates mitochondrial protein synthesis by deacetylation of the
ribosomal protein MRPL10, J. Biol. Chem., 285, 7417-7429,
doi: 10.1074/jbc.M109.053421.
20.Hirschey, M. D., Shimazu, T., Goetzman, E., Jing,
E., Schwer, B., et al. (2010) SIRT3 regulates mitochondrial fatty-acid
oxidation by reversible enzyme deacetylation, Nature,
464, 121-125, doi: 10.1038/nature08778.
21.Xue, L., Xu, F., Meng, L., Wei, S., Wang, J., et
al. (2012) Acetylation-dependent regulation of mitochondrial ALDH2
activation by SIRT3 mediates acute ethanol-induced eNOS activation,
FEBS Lett., 586, 137-142, doi:
10.1016/j.febslet.2011.11.031.
22.Wang, Z., Inuzuka, H., Zhong, J., Liu, P.,
Sarkar, F. H., et al. (2012) Identification of acetylation-dependent
regulatory mechanisms that govern the oncogenic functions of Skp2,
Oncotarget, 3, 1294-1300, doi:
10.18632/oncotarget.740.
23.Tseng, A. H. H., Shieh, S. S., and Wang, D. L.
(2013) SIRT3 deacetylates FOXO3 to protect mitochondria against
oxidative damage, Free Radic. Biol. Med., 63, 222-234,
doi: 10.1016/j.freeradbiomed.2013.05.002.
24.Jing, E., O’Neill, B. T., Rardin, M. J.,
Kleinridders, A., Ilkeyeva, O. R., et al. (2013) Sirt3 regulates
metabolic flexibility of skeletal muscle through reversible enzymatic
deacetylation, Diabetes, 62, 3404-3417, doi:
10.2337/db12-1650.
25.Cheng, Y., Ren, X., Gowda, A. S. P., Shan, Y.,
Zhang, L., et al. (2013) Interaction of Sirt3 with OGG1 contributes to
repair of mitochondrial DNA and protects from apoptotic cell death
under oxidative stress, Cell Death Dis., 4, 1-11, doi:
10.1038/cddis.2013.254.
26.Samant, S. A., Zhang, H. J., Hong, Z., Pillai, V.
B., Sundaresan, N. R., et al. (2014) SIRT3 deacetylates and activates
OPA1 to regulate mitochondrial dynamics during stress, Mol. Cell.
Biol., 3, 807-819, doi: 10.1128/mcb.01483-13.
27.Lu, Z., Chen, Y., Aponte, A. M., Battaglia, V.,
Gucek, M., et al. (2015) Prolonged fasting identifies heat shock
protein 10 as a sirtuin 3 substrate: Elucidating a new mechanism
linking mitochondrial protein acetylation to fatty acid oxidation
enzyme folding and function, J. Biol. Chem., 290,
2466-2476, doi: 10.1074/jbc.M114.606228.
28.Rauh, D., Fischer, F., Gertz, M.,
Lakshminarasimhan, M., Bergbrede, T., et al. (2013) An acetylome
peptide microarray reveals specificities and deacetylation substrates
for all human sirtuin isoforms, Nat. Commun., 4,
2327-2337, doi: 10.1038/ncomms3327.
29.Yang, H., Zhou, L., Shi, Q., Zhao, Y., Lin, H.,
et al. (2015) SIRT 3‐dependent GOT2 acetylation status affects
the malate-aspartate NADH shuttle activity and pancreatic tumor growth,
EMBO J., 34, 1110-1125, doi: 10.15252/embj.201591041.
30.Rardin, M. J., Newman, J. C., Held, J. M.,
Cusack, M. P., Sorensen, D. J., et al. (2013) Label-free quantitative
proteomics of the lysine acetylome in mitochondria identifies
substrates of SIRT3 in metabolic pathways, Proc. Natl. Acad. Sci.
USA, 110, 6601-6606, doi: 10.1073/pnas.1302961110.
31.Hebert, A. S., Dittenhafer-Reed, K. E., Yu, W.,
Bailey, D. J., Selen, E. S., et al. (2013) Calorie restriction and
SIRT3 trigger global reprogramming of the mitochondrial protein
acetylome, Mol. Cell., 49, 186-199, doi:
10.1016/j.molcel.2012.10.024.
32.Sol, E. M., Wagner, S. A., Weinert, B. T., Kumar,
A., Kim, H. S., et al. (2012) Proteomic investigations of lysine
acetylation identify diverse substrates of mitochondrial deacetylase
Sirt3, PLoS One, 7, 1-9, doi:
10.1371/journal.pone.0050545.
33.Consortium, T. U. (2021) UniProt: The universal
protein knowledgebase in 2021, Nucleic Acids Res.,
49, D480-D489, doi: 10.1093/nar/gkaa1100.
34.Amberger, J. S., Bocchini, C. A., Scott, A. F.,
and Hamosh, A. (2019) OMIM. org: Leveraging knowledge across
phenotype-gene relationships, Nucleic Acids Res., 47,
D1038-D1043, doi: 10.1093/nar/gky1151.
35.Zuberi, K., Franz, M., Rodriguez, H., Montojo,
J., Lopes, C. T., et al. (2013) GeneMANIA prediction server 2013
update, Nucleic Acids Res., 41, 115-122, doi:
10.1093/nar/gkt533.
36.Warde-Farley, D., Donaldson, S. L., Comes, O.,
Zuberi, K., Badrawi, R., et al. (2010) The GeneMANIA prediction server:
Biological network integration for gene prioritization and predicting
gene function, Nucleic Acids Res., 38, 214-220, doi:
10.1093/nar/gkq537.
37.Franz, M., Rodriguez, H., Lopes, C., Zuberi, K.,
Montojo, J., et al. (2018) GeneMANIA update 2018, Nucleic Acids
Res., 46, W60-W64, doi: 10.1093/nar/gky311.
38.Lopes, C. T., Franz, M., Kazi, F., Donaldson, S.
L., Morris, Q., et al. (2010) Cytoscape Web: An interactive web-based
network browser, Bioinformatics, 26, 2347-2348, doi:
10.1093/bioinformatics/btq430.
39.Szklarczyk, D., Gable, A. L., Lyon, D., Junge,
A., Wyder, S., et al. (2019) STRING v11: Protein–protein
association networks with increased coverage, supporting functional
discovery in genome-wide experimental datasets, Nucleic Acids
Res., 47, D607-D613, doi: 10.1093/nar/gky1131.
40.Enright, A. J., Van Dongen, S., and Ouzounis, C.
A. (2002) An efficient algorithm for large-scale detection of protein
families, Nucleic Acids Res., 30, 1575-1584, doi:
10.1093/nar/30.7.1575.
41.Chin, C.-H. H., Chen, S.-H. H., Wu, H.-H. H., Ho,
C.-W. W., Ko, M.-T. T., et al. (2014) CytoHubba: Identifying hub
objects and sub-networks from complex interactome, BMC Syst.
Biol., 8, 1-7, doi: 10.1186/1752-0509-8-S4-S11.
42.Bader, G. D., and Hogue, C. W. V. (2003) An
automated method for finding molecular complexes in large protein
interaction networks, BMC Bioinformatics, 4, 1-27, doi:
10.1186/1471-2105-4-2.
43.Kutmon, M., Lotia, S., Evelo, C. T., and Pico, A.
R. (2014) WikiPathways App for Cytoscape: Making biological pathways
amenable to network analysis and visualization, F1000Res.,
3, 152, doi: 10.12688/f1000research.4254.2.
44.Liu, L., Peritore, C., Ginsberg, J., Kayhan, M.,
and Donmez, G. (2015) SIRT3 attenuates MPTP-induced nigrostriatal
degeneration via enhancing mitochondrial antioxidant capacity,
Neurochem. Res., 40, 600-608, doi:
10.1007/s11064-014-1507-8.
45.Bader, J. S., Chaudhuri, A., Rothberg, J. M., and
Chant, J. (2004) Gaining confidence in high-throughput protein
interaction networks, Nat. Biotechnol., 22, 78-85, doi:
10.1038/nbt924.
46.Franceschini, A., Szklarczyk, D., Frankild, S.,
Kuhn, M., Simonovic, M., et al. (2013) STRING v9.1:
Protein–protein interaction networks, with increased coverage and
integration, Nucleic Acids Res., 41, 808-815, doi:
10.1093/nar/gks1094.
47.Szklarczyk, D., Morris, J. H., Cook, H., Kuhn,
M., Wyder, S., et al. (2017) The STRING database in 2017:
Quality-controlled protein–protein association networks, made
broadly accessible, Nucleic Acids Res., 45, D362-D368,
doi: 10.1093/nar/gkw937.
48.Van Dongen, S. M. (2000) Graph Clustering by
Flow Simulation, Utrecht University Repository. Dissertation
[English]. Utrecht University, Utrecht.
49.Ahn, B. H., Kim, H. S., Song, S., In, H. L., Liu,
J., et al. (2008) A role for the mitochondrial deacetylase Sirt3 in
regulating energy homeostasis, Proc. Natl. Acad. Sci. USA,
105, 14447-14452, doi: 10.1073/pnas.0803790105.
50.Fujino, T., Kondo, J., Ishikawa, M., Morikawa,
K., and Yamamoto, T. T. (2001) Acetyl-CoA Synthetase 2, a mitochondrial
matrix enzyme involved in the oxidation of acetate, J. Biol.
Chem., 276, 11420-11426, doi: 10.1074/jbc.M008782200.
51.Bao, J., Scott, I., Lu, Z., Pang, L., Dimond, C.
C., et al. (2010) SIRT3 is regulated by nutrient excess and modulates
hepatic susceptibility to lipotoxicity, Free Radic. Biol. Med.,
49, 1230-1237, doi: 10.1016/j.freeradbiomed.2010.07.009.
52.Finley, L. W. S., Haas, W., Desquiret-Dumas, V.,
Wallace, D. C., Procaccio, V., et al. (2011) Succinate dehydrogenase is
a direct target of sirtuin 3 deacetylase activity, PLoS One,
6, 4-9, doi: 10.1371/journal.pone.0023295.
53.Cimen, H., Han, M.-J., Yang, Y., Tong, Q., Koc,
H., et al. (2010) Regulation of succinate dehydrogenase activity by
SIRT3 in mammalian mitochondria, Biochemistry, 49,
304-311, doi: 10.1021/bi901627u.
54.Yang, W., Nagasawa, K., Münch, C., Xu, Y.,
Satterstrom, K., et al. (2016) Mitochondrial sirtuin network reveals
dynamic SIRT3-dependent deacetylation in response to membrane
depolarization, Cell, 167, 985-1000, e21, doi:
10.1016/j.cell.2016.10.016.
55.Bubber, P., Haroutunian, V., Fisch, G., Blass, J.
P., and Gibson, G. E. (2005) Mitochondrial abnormalities in Alzheimer
brain: Mechanistic implications, Ann. Neurol., 57,
695-703, doi: 10.1002/ana.20474.
56.Ozden, O., Park, S.-H., Wagner, B. A., Song, H.
Y., Zhu, Y., et al. (2014) SIRT3 deacetylates and increases pyruvate
dehydrogenase activity in cancer cells, Free Radic. Biol. Med.,
76, 163-172, doi: 10.1016/j.freeradbiomed.2014.08.001.
57.Warburg, O., Wind, F., Negelein, E., and Shirlaw,
J. T. (1927) The metabolism of tumors in the body, J. Gen.
Physiol., 8, 519-530, doi: 10.1085/jgp.8.6.519.
58.Stacpoole, P. W. (2012) The pyruvate
dehydrogenase complex as a therapeutic target for age-related diseases,
Aging Cell, 11, 371-377, doi:
10.1111/j.1474-9726.2012.00805.x.
59.Zhou, Q., Lam, P. Y., Han, D., and Cadenas, E.
(2009) Activation of c-Jun-N-terminal kinase and decline of
mitochondrial pyruvate dehydrogenase activity during brain aging,
FEBS Lett., 583, 1132-1140, doi:
10.1016/j.febslet.2009.02.043.
60.Scher, M. B., Vaquero, A., and Reinberg, D.
(2007) SirT3 is a nuclear NAD+-dependent histone deacetylase
that translocates to the mitochondria upon cellular stress, Genes
Dev., 21, 920-928, doi: 10.1101/gad.1527307.
61.Cooper, H. M., and Spelbrink, J. N. (2008) The
human SIRT3 protein deacetylase is exclusively mitochondrial,
Biochem. J., 411, 279-285, doi: 10.1042/BJ20071624.
62.Gurd, B. J., Holloway, G. P., Yoshida, Y., and
Bonen, A. (2012) In mammalian muscle, SIRT3 is present in mitochondria
and not in the nucleus; and SIRT3 is upregulated by chronic muscle
contraction in an adenosine monophosphate-activated protein
kinase-independent manner, Metabolism, 61, 733-741, doi:
10.1016/j.metabol.2011.09.016.
63.Vedrenne, V., Gowher, A., De Lonlay, P.,
Nitschke, P., Serre, V., et al. (2012) Mutation in PNPT1, which encodes
a Polyribonucleotide Nucleotidyltransferase, impairs RNA import into
mitochondria and causes respiratory-chain deficiency, Am. J. Hum.
Genet., 91, 912-918, doi: 10.1016/j.ajhg.2012.09.001.
64.Palmieri, L., Pardo, B., Lasorsa, F. M., del
Arco, A., Kobayashi, K., et al. (2001) Citrin and aralar1 are
Ca2+-stimulated aspartate/glutamate transporters in
mitochondria, EMBO J., 20, 5060-5069, doi:
10.1093/emboj/20.18.5060.
65.Galmiche, L., Serre, V., Beinat, M., Zossou, R.,
Assouline, Z., et al. (2012) Toward genotype phenotype correlations in
GFM1 mutations, Mitochondrion, 12, 242-247.
66.Fukumura, S., Ohba, C., Watanabe, T., Minagawa,
K., Shimura, M., et al. (2015) Compound heterozygous GFM2 mutations
with Leigh syndrome complicated by arthrogryposis multiplex congenita,
J. Hum. Genet., 60, 509-513, doi:
10.1038/jhg.2015.57.
67.Perli, E., Pisano, A., Glasgow, R. I. C., Carbo,
M., Hardy, S. A., et al. (2019) Novel compound mutations in the
mitochondrial translation elongation factor (TSFM) gene cause severe
cardiomyopathy with myocardial fibro-adipose replacement, Sci.
Rep., 9, 1-13, doi: 10.1038/s41598-019-41483-9.
68.Shi, H., Hayes, M., Kirana, C., Miller, R.,
Keating, J., et al. (2012) TUFM is a potential new prognostic indicator
for colorectal carcinoma, Pathology, 44, 506-512, doi:
10.1097/PAT.0b013e3283559cbe.
69.Sengupta, A., and Haldar, D. (2018) Human sirtuin
3 (SIRT3) deacetylates histone H3 lysine 56 to promote nonhomologous
end-joining repair, DNA Repair (Amst)., 61, 1-16, doi:
10.1016/j.dnarep.2017.11.003.
70.Nakamura, Y., Ogura, M., Tanaka, D., and Inagaki,
N. (2008) Localization of mouse mitochondrial SIRT proteins: Shift of
SIRT3 to the nucleus by co-expression with SIRT5, Biochem. Biophys.
Res. Commun., 366, 174-179, doi:
10.1016/j.bbrc.2007.11.122.
71.Redwood, A. B., Perkins, S. M., Vanderwaal, R.
P., Feng, Z., Biehl, K. J., et al. (2011) A dual role for A-type lamins
in DNA double-strand break repair, Cell Cycle, 10,
2549-2560, doi: 10.4161/cc.10.15.16531.
72.Murray-Nerger, L. A., Justice, J. L., Rekapalli,
P., Hutton, J. E., and Cristea, I. M. M. (2021) Lamin B1 acetylation
slows the G1 to S cell cycle transition through inhibition of DNA
repair, Nucleic Acids Res., 49, 2044-2064, doi:
10.1093/nar/gkab019.
73.Maynard, S., Keijzers, G., Akbari, M., Ezra, M.
B., Hall, A., et al. (2019) Lamin A/C promotes DNA base excision
repair, Nucleic Acids Res., 47, 11709-11728, doi:
10.1093/nar/gkz912.
74.Laemmle, A., Lechleiter, A., Roh, V., Schwarz,
C., Portmann, S., et al. (2012) Inhibition of SIRT1 impairs the
accumulation and transcriptional activity of HIF-1α protein under
hypoxic conditions, PLoS One, 7, e33433, doi:
10.1371/journal.pone.0033433.
75.Joo, H.-Y., Yun, M., Jeong, J., Park, E.-R.,
Shin, H.-J., et al. (2015) SIRT1 deacetylates and stabilizes
hypoxia-inducible factor-1α (HIF-1α) via direct
interactions during hypoxia, Biochem. Biophys. Res. Commun.,
462, 294-300, doi: 10.1016/j.bbrc.2015.04.119.
76.Xiong, Y., Wang, L., Wang, S., Wang, M., Zhao,
J., et al. (2018) SIRT3 deacetylates and promotes degradation of P53 in
PTEN-defective non-small cell lung cancer, J. Cancer Res. Clin.
Oncol., 144, 189-198, doi: 10.1007/s00432-017-2537-9.
77.Lee, J., Kim, Y., Liu, T., Hwang, Y. J., Hyeon,
S. J., et al. (2018) SIRT3 deregulation is linked to mitochondrial
dysfunction in Alzheimer’s disease, Aging Cell, 17,
1-12, doi: 10.1111/acel.12679.
78.Polischouk, A. G., Cedervall, B., Ljungquist, S.,
Flygare, J., Hellgren, D., et al. (1999) DNA double-strand break
repair, DNA-PK, and DNA ligases in two human squamous carcinoma cell
lines with different radiosensitivity, Int. J. Radiat. Oncol.,
43, 191-198, doi: 10.1016/S0360-3016(98)00362-9.
79.Chaplin, A. K., Hardwick, S. W., Liang, S.,
Kefala Stavridi, A., Hnizda, A., et al. (2021) Dimers of DNA-PK create
a stage for DNA double-strand break repair, Nat. Struct. Mol.
Biol., 28, 13-19, doi: 10.1038/s41594-020-00517-x.
80.Park, S. J., Gavrilova, O., Brown, A. L., Soto,
J. E., Bremner, S., et al. (2017) DNA-PK promotes the mitochondrial,
metabolic, and physical decline that occurs during aging, Cell
Metab., 25, 1135-1146.e7, doi:
10.1016/j.cmet.2017.04.008.
81.Sui, J., Zhang, S., and Chen, B. P. C. (2020)
DNA-dependent protein kinase in telomere maintenance and protection,
Cell. Mol. Biol. Lett., 25, 1-14, doi:
10.1186/s11658-020-0199-0.
82.Schrader, M., Kamoshita, M., and Islinger, M.
(2020) Organelle interplay-peroxisome interactions in health and
disease, J. Inherit. Metab. Dis., 43, 71-89, doi:
10.1002/jimd.12083.
83.Palacios, O. M., Carmona, J. J., Michan, S.,
Chen, K. Y., Manabe, Y., et al. (2009) Diet and exercise signals
regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle,
Aging (Albany NY), 1, 771-783, doi:
10.18632/aging.100075.
84.Ramesh, S., Govindarajulu, M., Lynd, T., Briggs,
G., Adamek, D., et al. (2018) SIRT3 activator Honokiol attenuates
β-amyloid by modulating amyloidogenic pathway, PLoS One,
13, 1, doi: 10.1371/journal.pone.0190350.
85.Shi, T., Wang, F., Stieren, E., and Tong, Q.
(2005) SIRT3, a mitochondrial sirtuin deacetylase, regulates
mitochondrial function and thermogenesis in brown adipocytes, J.
Biol. Chem., 280, 13560-13567, doi:
10.1074/jbc.M414670200.
86.Katsouri, L., Parr, C., Bogdanovic, N., Willem,
M., and Sastre, M. (2011) PPARγ co-activator-1α
(PGC-1α) reduces amyloid-β generation through a
PPARγ-dependent mechanism, J. Alzheimer’s Dis.,
25, 151-162, doi: 10.3233/JAD-2011-101356.
87.Liu, C., Li, S., Liu, T., Borjigin, J., and Lin,
J. D. (2007) Transcriptional coactivator PGC-1α integrates the
mammalian clock and energy metabolism, Nature, 447,
477-481, doi: 10.1038/nature05767.
88.Song, C., Zhao, J., Zhang, J., Mao, T., Fu, B.,
et al. (2017) SIRT3-dependent mitochondrial oxidative stress in sodium
fluoride-induced hepatotoxicity and salvage by melatonin,
BioRxiv, doi: 10.1101/107813.
89.Mauvoisin, D., Atger, F., Dayon, L.,
Núñez Galindo, A., Wang, J., et al. (2017) Circadian and
feeding rhythms orchestrate the diurnal liver acetylome, Cell
Rep., 20, 1729-1743, doi: 10.1016/j.celrep.2017.07.065.
90.Kondratova, A. A., and Kondratov, R. V. (2012)
The circadian clock and pathology of the ageing brain, Nat. Rev.
Neurosci., 13, 325-335, doi: 10.1038/nrn3208.
91.Tomita, T. (2017) Aberrant proteolytic processing
and therapeutic strategies in Alzheimer’s disease, Adv. Biol.
Regul., 64, 33-38, doi: 10.1016/j.jbior.2017.01.001.
92.Chaves, I., van der Horst, G. T. J., Schellevis,
R., Nijman, R. M., Koerkamp, M. G., et al. (2014) Insulin-FOXO3
signaling modulates circadian rhythms via regulation of clock
transcription, Curr. Biol., 24, 1248-1255, doi:
10.1016/j.cub.2014.04.018.
93.Tong, W., Ju, L., Qiu, M., Xie, Q., Chen, Y., et
al. (2016) Liraglutide ameliorates non-alcoholic fatty liver disease by
enhancing mitochondrial architecture and promoting autophagy through
the SIRT1/SIRT3-FOXO3a pathway, Hepatol. Res., 46,
933-943, doi: 10.1111/hepr.12634.
94.Jacobs, K. M., Pennington, J. D., Bisht, K. S.,
Aykin-Burns, N., Kim, H.-S., et al. (2008) SIRT3 interacts with the
daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a
dependent gene expression, Int. J. Biol. Sci., 4,
4291-4299, doi: 10.7150/ijbs.4.291.
Supplementary Table S1 (PDF)
Supplementary Table S2 (PDF)